Coherent Exciton Dynamics in Ensembles of Size-Dispersed CdSe Quantum Dot Dimers Probed via Ultrafast Spectroscopy: A Quantum Computational Study



Abstract
Featured Application
Abstract
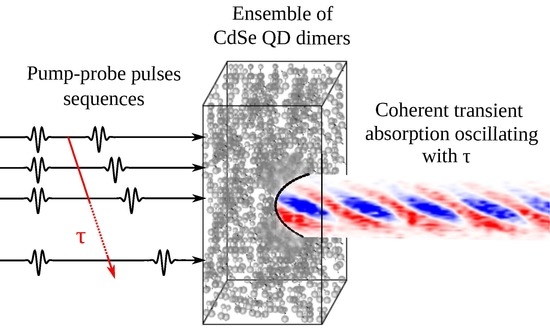
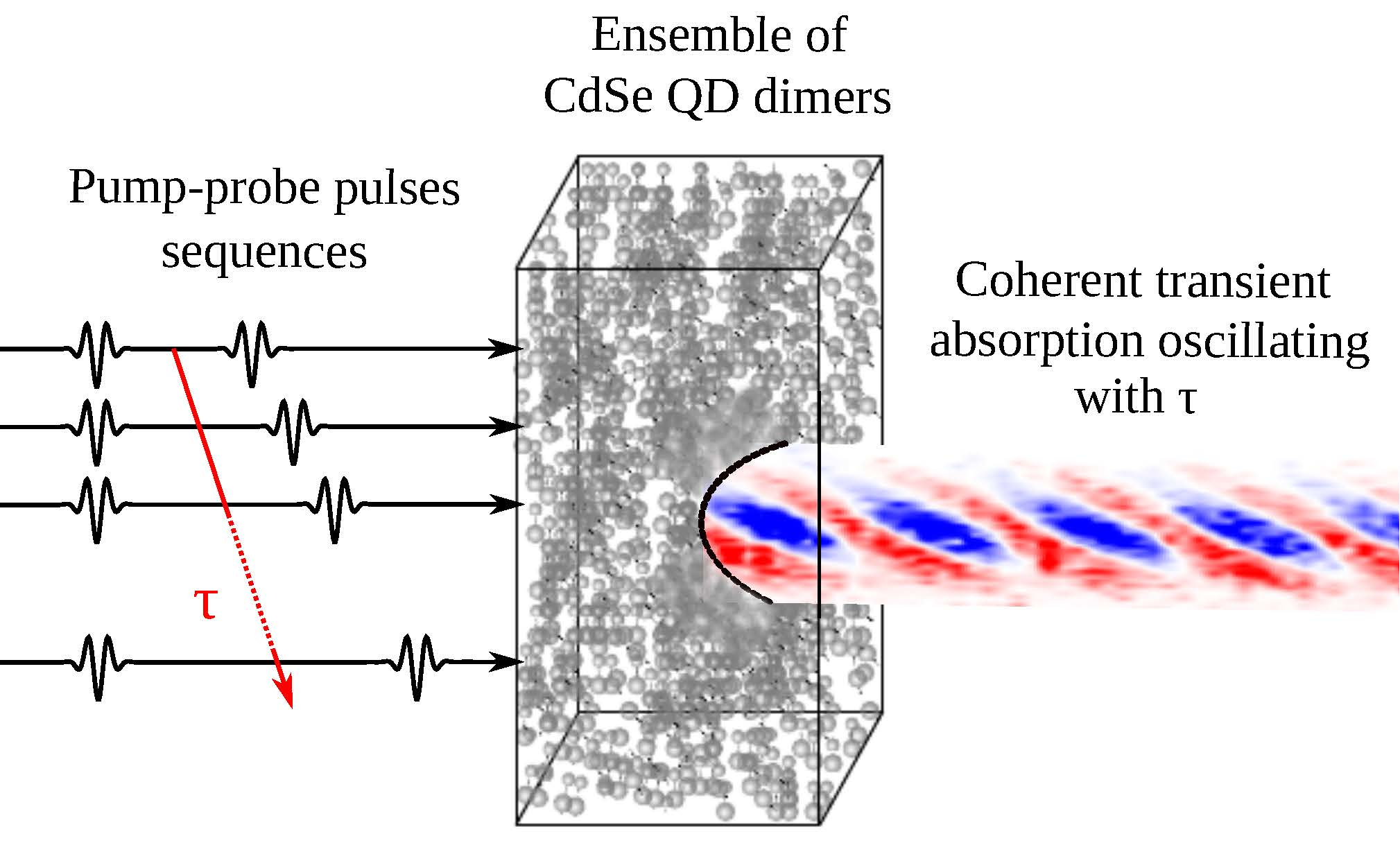{kind=link}
{kind=link}
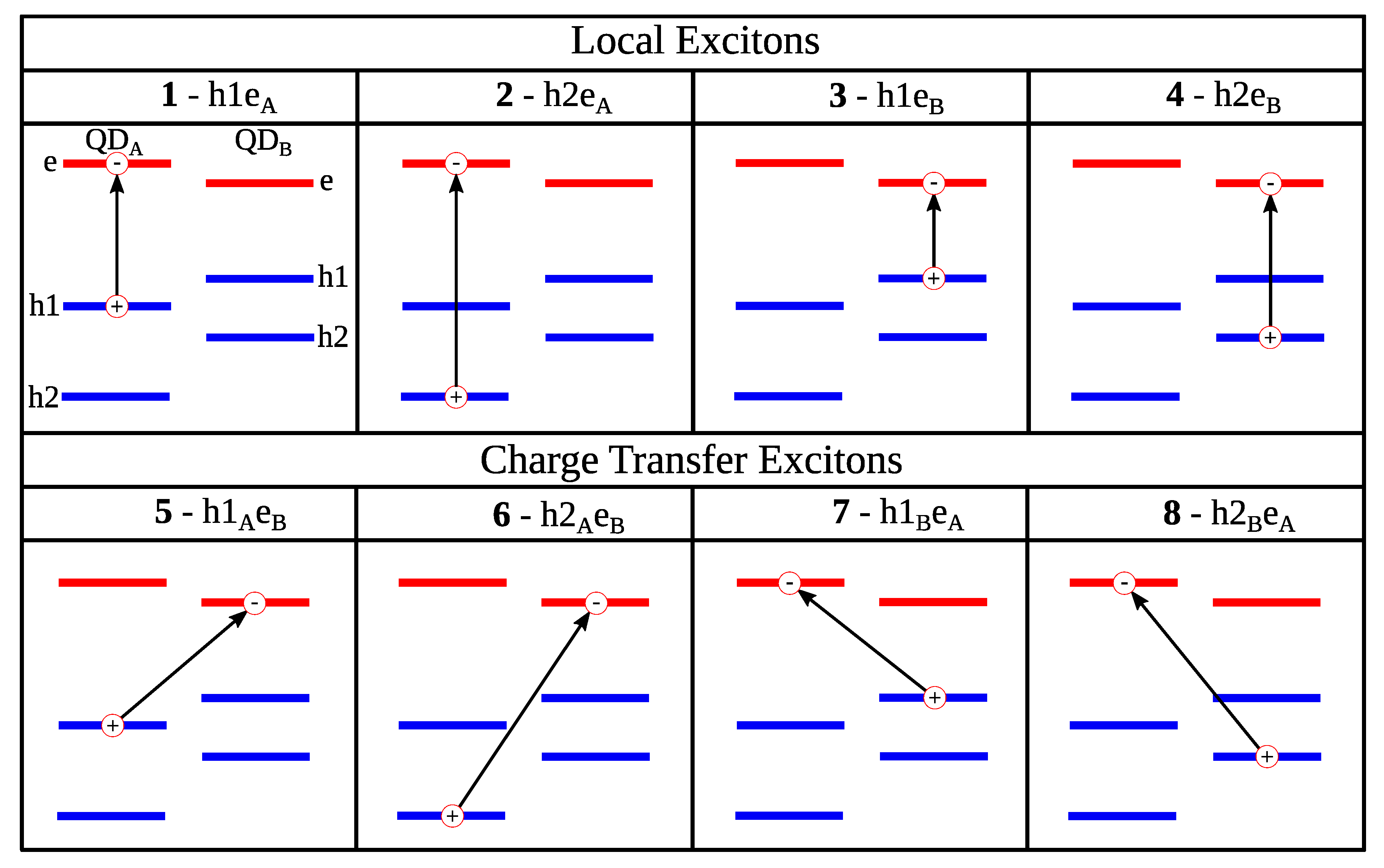{kind=link}
{kind=link}
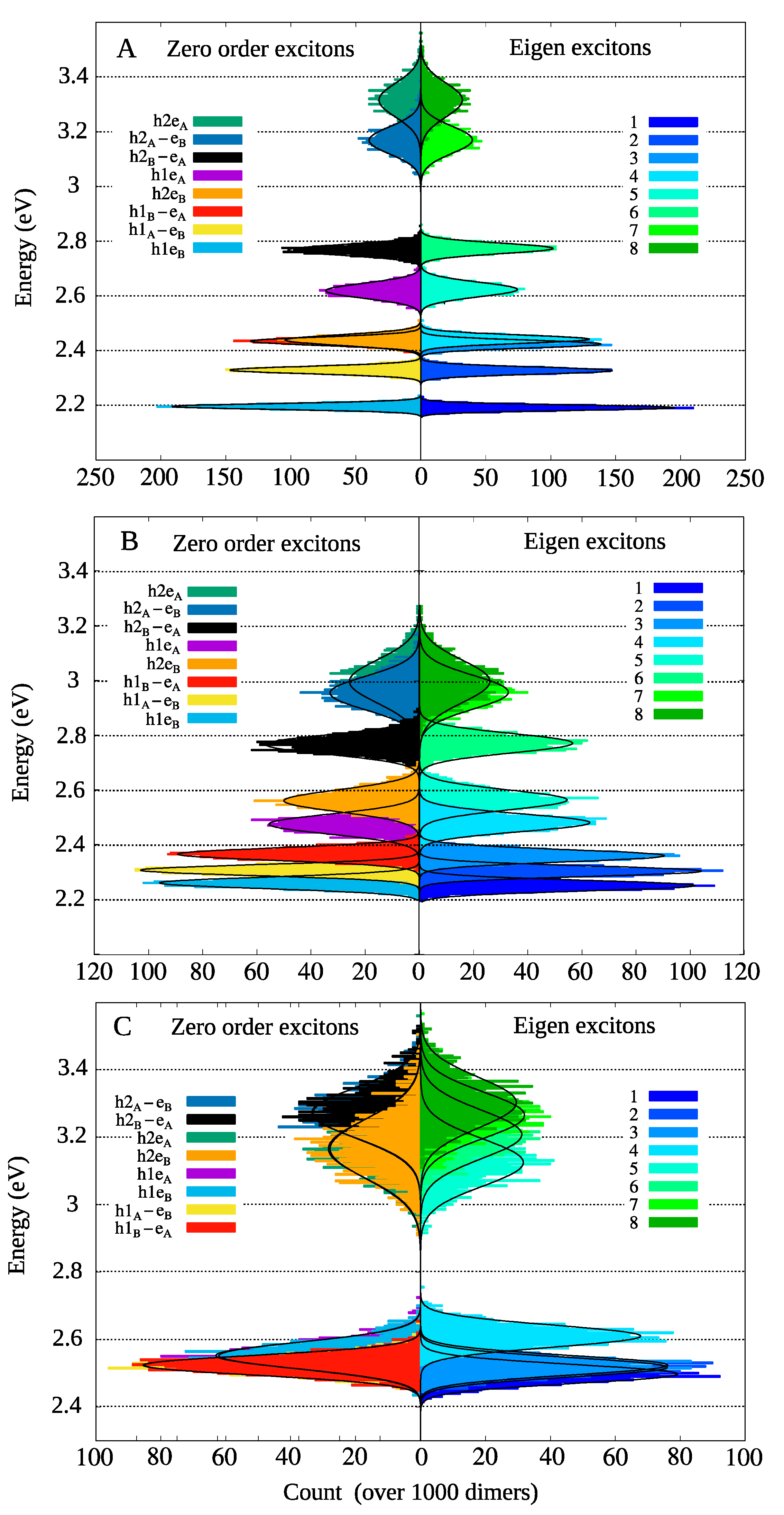{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods: Model Excitonic Hamiltonian
3. Results
3.1. Effect of the QD Size Dispersion on Excitonic Level Structure
3.2. Coherent Excitonic Quantum Electronic Dynamics in the QD Dimer
4. Discussion: Probing the Coherent Intradot and Interdot Dynamics in Dimer Ensembles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alivisatos, A.P. Semiconductor Clusters, Nanocrystals, and Quantum Dots. Science 1996, 271, 933. [Google Scholar] [CrossRef]
- Markovich, G.; Collier, C.P.; Henrichs, S.E.; Remacle, F.; Levine, R.D.; Heath, J.R. Architectonic Quantum Dot Solids. Acc. Chem. Res. 1999, 32, 415–423. [Google Scholar] [CrossRef]
- Whetten, R.L.; Shafigullin, M.N.; Khoury, J.T.; Schaaff, T.G.; Vezmar, I.; Alvarez, M.M.; Wilkinson, A. Crystal Structures of Molecular Gold Nanocrystal Arrays. Acc. Chem. Res. 1999, 32, 397–406. [Google Scholar] [CrossRef]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Synthesis and Characterization of Monodisperse Nanocrystals and Close-Packed Nanocrystal Assemblies. Ann. Rev. Mater. Sci. 2000, 30, 545–610. [Google Scholar] [CrossRef]
- Remacle, F.; Levine, R.D. Architecture with Designer Atoms: Simple Theoretical Considerations. Proc. Natl. Acad. Sci. USA 2000, 97, 553–558. [Google Scholar] [CrossRef]
- Nanocrystal Quantum Dots, 2nd ed.; Klimov, V.I., Ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Xu, X.; Stöttinger, S.; Battagliarin, G.; Hinze, G.; Mugnaioli, E.; Li, C.; Müllen, K.; Basché, T. Assembly and Separation of Semiconductor Quantum Dot Dimers and Trimers. J. Am. Chem. Soc. 2011, 133, 18062–18065. [Google Scholar] [CrossRef]
- Williams, K.J.; Tisdale, W.A.; Leschkies, K.S.; Haugstad, G.; Norris, D.J.; Aydil, E.S.; Zhu, X.Y. Strong Electronic Coupling in Two-Dimensional Assemblies of Colloidal PbSe Quantum Dots. ACS Nano 2009, 3, 1532–1538. [Google Scholar] [CrossRef]
- Koole, R.; Liljeroth, P.; de Mello Donegá, C.; Vanmaekelbergh, D.; Meijerink, A. Electronic Coupling and Exciton Energy Transfer in CdTe Quantum-Dot Molecules. J. Am. Chem. Soc. 2006, 128, 10436–10441. [Google Scholar] [CrossRef]
- Cohen, E.; Komm, P.; Rosenthal-Strauss, N.; Dehnel, J.; Lifshitz, E.; Yochelis, S.; Levine, R.D.; Remacle, F.; Fresch, B.; Marcus, G.; et al. Fast Energy Transfer in CdSe Quantum Dot Layered Structures: Controlling Coupling with Covalent-Bond Organic Linkers. J. Phys. Chem. C 2018, 122, 5753–5758. [Google Scholar] [CrossRef]
- Grimaldi, G.; Crisp, R.W.; ten Brinck, S.; Zapata, F.; van Ouwendorp, M.; Renaud, N.; Kirkwood, N.; Evers, W.H.; Kinge, S.; Infante, I.; et al. Hot-electron transfer in quantum-dot heterojunction films. Nat. Commun. 2018, 9, 2310. [Google Scholar] [CrossRef]
- Kagan, C.R.; Lifshitz, E.; Sargent, E.H.; Talapin, D.V. Building devices from colloidal quantum dots. Science 2016, 353, aac5523. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Lee, J.-S.; Kovalenko, M.V.; Shevchenko, E.V. Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications. Chem. Rev. 2010, 110, 389–458. [Google Scholar] [CrossRef] [PubMed]
- Panfil, Y.E.; Oded, M.; Banin, U. Colloidal Quantum Nanostructures: Emerging Materials for Display Applications. Angew. Chem. Int. Ed. 2018, 57, 4274–4295. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.B.; Norris, D.J.; Bawendi, M.G. Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 1993, 115, 8706–8715. [Google Scholar] [CrossRef]
- Kagan, C.R.; Murray, C.B.; Nirmal, M.; Bawendi, M.G. Electronic Energy Transfer in CdSe Quantum Dot Solids. Phys. Rev. Lett. 1996, 76, 1517–1520. [Google Scholar] [CrossRef]
- Norris, D.J.; Bawendi, M.G. Measurement and assignment of the size-dependent optical spectrum in CdSe quantum dots. Phys. Rev. B 1996, 53, 16338–16346. [Google Scholar] [CrossRef]
- Efros, A.L.; Rosen, M.; Kuno, M.; Nirmal, M.; Norris, D.J.; Bawendi, M. Band-edge exciton in quantum dots of semiconductors with a degenerate valence band: Dark and bright exciton states. Phys. Rev. B 1996, 54, 4843–4856. [Google Scholar] [CrossRef]
- Soloviev, V.N.; Eichhöfer, A.; Fenske, D.; Banin, U. Size-Dependent Optical Spectroscopy of a Homologous Series of CdSe Cluster Molecules. J. Am. Chem. Soc. 2001, 123, 2354–2364. [Google Scholar] [CrossRef]
- Sercel, P.C.; Efros, A.L. Band-Edge Exciton in CdSe and Other II–VI and III–V Compound Semiconductor Nanocrystals—Revisited. Nano Lett. 2018, 18, 4061–4068. [Google Scholar] [CrossRef]
- Sercel, P.C.; Shabaev, A.; Efros, A.L. Photoluminescence Enhancement through Symmetry Breaking Induced by Defects in Nanocrystals. Nano Lett. 2017, 17, 4820–4830. [Google Scholar] [CrossRef]
- Ma, H.; Jin, Z.; Zhang, Z.; Li, G.; Ma, G. Exciton Spin Relaxation in Colloidal CdSe Quantum Dots at Room Temperature. J. Phys. Chem. A 2012, 116, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Moreels, I.; Rainò, G.; Gomes, R.; Hens, Z.; Stöferle, T.; Mahrt, R.F. Band-Edge Exciton Fine Structure of Small, Nearly Spherical Colloidal CdSe/ZnS Quantum Dots. ACS Nano 2011, 5, 8033–8039. [Google Scholar] [CrossRef] [PubMed]
- Huxter, V.M.; Scholes, G.D. Acoustic phonon strain induced mixing of the fine structure levels in colloidal CdSe quantum dots observed by a polarization grating technique. J. Chem. Phys. 2010, 132, 104506. [Google Scholar] [CrossRef] [PubMed]
- Caram, J.R.; Zheng, H.; Dahlberg, P.D.; Rolczynski, B.S.; Griffin, G.B.; Fidler, A.F.; Dolzhnikov, D.S.; Talapin, D.V.; Engel, G.S. Persistent Interexcitonic Quantum Coherence in CdSe Quantum Dots. J. Phys. Chem. Lett. 2014, 5, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Caram, J.R.; Zheng, H.; Dahlberg, P.D.; Rolczynski, B.S.; Griffin, G.B.; Dolzhnikov, D.S.; Talapin, D.V.; Engel, G.S. Exploring size and state dynamics in CdSe quantum dots using two-dimensional electronic spectroscopy. J. Chem. Phys. 2014, 140, 084701. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Trivedi, D.; Chakrabortty, S.; Kobayashi, T.; Chan, Y.; Prezhdo, O.V.; Loh, Z.-H. Observation of an Excitonic Quantum Coherence in CdSe Nanocrystals. Nano Lett. 2015, 15, 6875–6882. [Google Scholar] [CrossRef]
- Janke, E.M.; Williams, N.E.; She, C.; Zherebetskyy, D.; Hudson, M.H.; Wang, L.; Gosztola, D.J.; Schaller, R.D.; Lee, B.; Sun, C.; et al. Origin of Broad Emission Spectra in InP Quantum Dots: Contributions from Structural and Electronic Disorder. J. Am. Chem. Soc. 2018, 140, 15791–15803. [Google Scholar] [CrossRef]
- Cassette, E.; Pensack, R.D.; Mahler, B.; Scholes, G.D. Room-temperature exciton coherence and dephasing in two-dimensional nanostructures. Nat. Commun. 2015, 6, 6086. Available online: https://www.nature.com/articles/ncomms7086#supplementary-information (accessed on 15 January 2015). [CrossRef]
- Wong, C.Y.; Scholes, G.D. Biexcitonic Fine Structure of CdSe Nanocrystals Probed by Polarization-Dependent Two-Dimensional Photon Echo Spectroscopy. J. Phys. Chem. A 2011, 115, 3797–3806. [Google Scholar] [CrossRef]
- Turner, D.B.; Hassan, Y.; Scholes, G.D. Exciton Superposition States in CdSe Nanocrystals Measured Using Broadband Two-Dimensional Electronic Spectroscopy. Nano Lett. 2012, 12, 880–886. [Google Scholar] [CrossRef]
- Collini, E.; Gattuso, H.; Bolzonello, L.; Casotto, A.; Volpato, A.; Dibenedetto, C.N.; Fanizza, E.; Striccoli, M.; Remacle, F. Quantum Phenomena in Nanomaterials: Coherent Superpositions of Fine Structure States in CdSe Nanocrystals at Room Temperature. J. Phys. Chem. C 2019. [Google Scholar] [CrossRef]
- Klimov, V.I.; McBranch, D.W.; Leatherdale, C.A.; Bawendi, M.G. Electron and hole relaxation pathways in semiconductor quantum dots. Phys. Rev. B 1999, 60, 13740–13749. [Google Scholar] [CrossRef]
- Klimov, V.I. Mechanisms for Photogeneration and Recombination of Multiexcitons in Semiconductor Nanocrystals: Implications for Lasing and Solar Energy Conversion. J. Phys. B 2006, 110, 16827–16845. [Google Scholar]
- Mlinar, V.; Zunger, A. Internal electronic structure and fine structure of multiexcitons in semiconductor quantum dots. Phys. Rev. B 2009, 80, 205311. [Google Scholar] [CrossRef]
- Sewall, S.L.; Franceschetti, A.; Cooney, R.R.; Zunger, A. Direct observation of the structure of band-edge biexcitons in colloidal semiconductor CdSe quantum dots. Phys. Rev. B 2009, 80, 081310. [Google Scholar] [CrossRef]
- Trivedi, D.J.; Wang, L.; Prezhdo, O.V. Auger-Mediated Electron Relaxation Is Robust to Deep Hole Traps: Time-Domain Ab Initio Study of CdSe Quantum Dots. Nano Lett. 2015, 15, 2086–2091. [Google Scholar] [CrossRef]
- Morgan, N.Y.; Leatherdale, C.A.; Drndić, M.; Jarosz, M.V.; Kastner, M.A.; Bawendi, M. Electronic transport in films of colloidal CdSe nanocrystals. Phys. Rev. B 2002, 66, 075339. [Google Scholar] [CrossRef]
- Liang, Y.; Thorne, J.E.; Parkinson, B.A. Controlling the Electronic Coupling between CdSe Quantum Dots and Thiol Capping Ligands via pH and Ligand Selection. Langmuir 2012, 28, 11072–11077. [Google Scholar] [CrossRef]
- Ginger, D.S.; Greenham, N.C. Charge injection and transport in films of CdSe nanocrystals. J. Appl. Phys. 2000, 87, 1361–1368. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kovalenko, M.V.; Huang, J.; Chung, D.S.; Talapin, D.V. Band-like transport, high electron mobility and high photoconductivity in all-inorganic nanocrystal arrays. Nat. Nanotechnol. 2011, 6, 348. Available online: https://www.nature.com/articles/nnano.2011.46#supplementary-information (accessed on 24 April 2011). [CrossRef]
- Brus, L.E. Electron–electron and electron-hole interactions in small semiconductor crystallites: The size dependence of the lowest excited electronic state. J. Chem. Phys. 1984, 80, 4403–4409. [Google Scholar] [CrossRef]
- Wang, Y.; Herron, N. Nanometer-sized semiconductor clusters: Materials synthesis, quantum size effects, and photophysical properties. J. Phys. Chem. 1991, 95, 525–532. [Google Scholar] [CrossRef]
- Koole, R.; Groeneveld, E.; Vanmaekelbergh, D.; Meijerink, A.; deMelloDonega, C. Size Effect on Semiconductor Nanoparticles. In Nanoparticles; deMelloDonega, C., Ed.; Springer: Berlin, Germany, 2014. [Google Scholar]
- Zunger, A. Electronic Structure Theory of Semiconductor Quantum Dots. MRS Bull. 1998, 23, 35–42. [Google Scholar] [CrossRef]
- Prezhdo, O.V. Photoinduced Dynamics in Semiconductor Quantum Dots: Insights from Time-Domain ab Initio Studies. Acc. Chem. Res. 2009, 42, 2005–2016. [Google Scholar] [CrossRef]
- Wang, L.-W.; Zunger, A. Pseudopotential calculations of nanoscale CdSe quantum dots. Phys. Rev. B 1996, 53, 9579–9582. [Google Scholar] [CrossRef]
- Wang, Z.A. High-Energy Excitonic Transitions in CdSe Quantum Dots. J. Phys. Chem. B 1998, 102, 6449–6454. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.-H.; Zhang, Y.; Kowalski, P.J.; Rohrs, H.W.; Buhro, W.E. Preparation of Primary Amine Derivatives of the Magic-Size Nanocluster (CdSe)13. Inorg. Chem. 2013, 52, 2933–2938. [Google Scholar] [CrossRef]
- Vogl, P.; Hjalmarson, H.P.; Dow, J.D. A semi-empirical tigh-binding theory of the electronic structure of semicoductors. J. Phys. Chem. Sol. 1983, 44, 365–378. [Google Scholar] [CrossRef]
- Klimeck, G.; Oyafuso, F.; Boykin, T.B.; Bowen, R.C.; Allmen, P.V. Development of a Nanoelectronic 3-D (NEMO 3-D) Simulator for Multimillion Atom Simulations and Its Application to Alloyed Quantum Dots. CMES 2002, 3, 602–642. [Google Scholar]
- Lee, S.; Jönsson, L.; Wilkins, J.W.; Bryant, G.W.; Klimeck, G. Electron-hole correlations in semiconductor quantum dots with tight-binding wave functions. Phys. Rev. B 2001, 63, 195318. [Google Scholar] [CrossRef]
- Kilina, S.V.; Kilin, D.S.; Prezhdo, O.V. Breaking the Phonon Bottleneck in PbSe and CdSe Quantum Dots: Time-Domain Density Functional Theory of Charge Carrier Relaxation. ACS Nano 2009, 3, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Trivedi, D.J.; Akimov, A.V.; Aradi, B.; Frauenheim, T.; Prezhdo, O.V. Nonadiabatic Molecular Dynamics for Thousand Atom Systems: A Tight-Binding Approach toward PYXAID. J. Chem. Theory Comput. 2016, 12, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Luttinger, J.M.; Kohn, W. Motion of Electrons and Holes in Perturbed Periodic Fields. Phys. Rev. 1955, 97, 869–883. [Google Scholar] [CrossRef]
- Brus, L. Electronic wave functions in semiconductor clusters: Experiment and theory. J. Phys. Chem. 1986, 90, 2555–2560. [Google Scholar] [CrossRef]
- Efros, A.L. Luminescence polarization of CdSe microcrystals. Phys. Rev. B 1992, 46, 7448–7458. [Google Scholar] [CrossRef]
- Efros, A.L.; Rosen, M. The electronic structure of semi-conducting nanocrystal. Ann. Rev. Mater. Sci. 2000, 30, 475–521. [Google Scholar] [CrossRef]
- Lovett, B.W.; Reina, J.H.; Nazir, A.; Briggs, G.A.D. Optical schemes for quantum computation in quantum dot molecules. Phys. Rev. B 2003, 68, 205319. [Google Scholar] [CrossRef]
- Nazir, A.; Lovett, B.W.; Barrett, S.D.; Reina, J.H.; Briggs, G.A.D. Anticrossings in F\orster coupled quantum dots. Phys. Rev. B 2005, 71, 045334. [Google Scholar] [CrossRef]
- Kruchinin, S.Y.; Fedorov, A.V.; Baranov, A.V.; Perova, T.S.; Berwick, K. Resonant energy transfer in quantum dots: Frequency-domain luminescent spectroscopy. Phys. Rev. B 2008, 78, 125311. [Google Scholar] [CrossRef]
- Kruchinin, S.Y.; Fedorov, A.V.; Baranov, A.V.; Perova, T.S.; Berwick, K. Electron-electron scattering in a double quantum dot: Effective mass approach. J. Chem. Phys. 2010, 133, 104704. [Google Scholar] [CrossRef]
- Troiani, F.; Hohenester, U.; Molinari, E. Electron-hole localization in coupled quantum dots. Phys. Rev. B 2002, 65, 161301. [Google Scholar] [CrossRef]
- Seibt, J.; Pullerits, T. Beating Signals in 2D Spectroscopy: Electronic or Nuclear Coherences? Application to a Quantum Dot Model System. J. Phys. Chem. C 2013, 117, 18728–18737. [Google Scholar] [CrossRef]
- Seibt, J.; Hansen, T.; Pullerits, T. 3D Spectroscopy of Vibrational Coherences in Quantum Dots: Theory. J. Phys. Chem. B 2013, 117, 11124–11133. [Google Scholar] [CrossRef] [PubMed]
- Karki, K.J.; Ma, F.; Zheng, K.; Zidek, K.; Mousa, A.; Abdellah, M.A.; Messing, M.E.; Wallenberg, L.R.; Yartsev, A.; Pullerits, T. Multiple exciton generation in nano-crystals revisited: Consistent calculation of the yield based on pump-probe spectroscopy. Sci. Rep. 2013, 3, 2287. [Google Scholar] [CrossRef]
- Wang, H.; de Mello Donegá, C.; Meijerink, A.; Glasbeek, M. Ultrafast Exciton Dynamics in CdSe Quantum Dots Studied from Bleaching Recovery and Fluorescence Transients. J. Phys. Chem. B 2006, 110, 733–737. [Google Scholar] [CrossRef]
- Kambhampati, P. Hot Exciton Relaxation Dynamics in Semiconductor Quantum Dots: Radiationless Transitions on the Nanoscale. J. Phys. Chem. C 2011, 115, 22089–22109. [Google Scholar] [CrossRef]
- Žídek, K.; Zheng, K.; Ponseca, C.S.; Messing, M.E.; Wallenberg, L.R.; Chábera, P.; Abdellah, M.; Sundström, V.; Pullerits, T. Electron Transfer in Quantum-Dot-Sensitized ZnO Nanowires: Ultrafast Time-Resolved Absorption and Terahertz Study. J. Am. Chem. Soc. 2012, 134, 12110–12117. [Google Scholar] [CrossRef]
- Walsh, B.R.; Sonnichsen, C.; Mack, T.G.; Saari, J.I.; Krause, M.M.; Nick, R.; Coe-Sullivan, S.; Kambhampati, P. Excited State Phononic Processes in Semiconductor Nanocrystals Revealed by Excitonic State-Resolved Pump/Probe Spectroscopy. J. Phys. Chem. C 2019, 123, 3868–3875. [Google Scholar] [CrossRef]
- Scholes, G.D. Coherence from Light Harvesting to Chemistry. J. Phys. Chem. Lett. 2018, 9, 1568–1572. [Google Scholar] [CrossRef]
- Fresch, B.; Cipolloni, M.; Yan, T.-M.; Collini, E.; Levine, R.D.; Remacle, F. Parallel and Multivalued Logic by the Two-Dimensional Photon-Echo Response of a Rhodamine–DNA Complex. J. Phys. Chem. Lett. 2015, 6, 1714–1718. [Google Scholar] [CrossRef]
- Fresch, B.; Hiluf, D.; Collini, E.; Levine, R.D.; Remacle, F. Molecular decision trees realized by ultrafast electronic spectroscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 17183–17188. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, L.-W.; Zunger, A. Excitonic exchange splitting in bulk semiconductors. Phys. Rev. B 1999, 59, 5568–5574. [Google Scholar] [CrossRef]
- Wang, L.-W.; Zunger, A. Local-density-derived semiempirical pseudopotentials. Phys. Rev. B 1995, 51, 17398–17416. [Google Scholar] [CrossRef] [PubMed]
- Kelley, A.M. Electron–Phonon Coupling in CdSe Nanocrystals from an Atomistic Phonon Model. ACS Nano 2011, 5, 5254–5262. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.S. Solid State Physics; Academic: New York, NY, USA, 1963; Volume 5. [Google Scholar]
- Franceschetti, A.; Fu, H.; Wang, L.W.; Zunger, A. Many-body pseudopotential theory of excitons in InP and CdSe quantum dots. Phys. Rev. B 1999, 60, 1819–1829. [Google Scholar] [CrossRef]
- Ferreyra, J.M.; Proetto, C.R. Quantum size effects on excitonic Coulomb and exchange energies in finite-barrier semiconductor quantum dots. Phys. Rev. B 1999, 60, 10672–10675. [Google Scholar] [CrossRef]
- Specht, J.F.; Knorr, A.; Richter, M. Two-dimensional spectroscopy: An approach to distinguish F\orster and Dexter transfer processes in coupled nanostructures. Phys. Rev. B 2015, 91, 155313. [Google Scholar] [CrossRef]
- Akimov, A.V.; Prezhdo, O.V. Large-Scale Computations in Chemistry: A Bird’s Eye View of a Vibrant Field. Chem. Rev. 2015, 115, 5797–5890. [Google Scholar] [CrossRef]
- Kelley, A.M. Electron−Phonon Coupling in CdSe Nanocrystals. J. Phys. Chem. Lett. 2010, 1, 1296–1300. [Google Scholar] [CrossRef]
- Salvador, M.R.; Graham, M.W.; Scholes, G.D. Exciton-phonon coupling and disorder in the excited states of CdSe colloidal quantum dots. J. Chem. Phys. 2006, 125, 184709. [Google Scholar] [CrossRef]
- Klimov, V.I. Optical Nonlinearities and Ultrafast Carrier Dynamics in Semiconductor Nanocrystals. J. Phys. Chem. B 2000, 104, 6112–6123. [Google Scholar] [CrossRef]
- Crooker, S.A.; Hollingsworth, J.A.; Tretiak, S.; Klimov, V.I. Spectrally Resolved Dynamics of Energy Transfer in Quantum-Dot Assemblies: Towards Engineered Energy Flows in Artificial Materials. Phys. Rev. Lett. 2002, 89, 186802. [Google Scholar] [CrossRef] [PubMed]
- Seidner, L.; Stock, G.; Domcke, W. Nonperturbative approach to femtosecond spectroscopy: General theory and application to multidimensional nonadiabatic photoisomerization processes. J. Chem. Phys. 1995, 103, 3998–4011. [Google Scholar] [CrossRef]
- Mengxi, W.; Shaohao, C.; Seth, C.; Kenneth, J.S.; Mette, B.G. Theory of strong-field attosecond transient absorption. J. Phys. B 2016, 49, 062003. [Google Scholar]
- Lin, C.; Gong, K.; Kelley, D.F.; Kelley, A.M. Size-Dependent Exciton–Phonon Coupling in CdSe Nanocrystals through Resonance Raman Excitation Profile Analysis. J. Phys. Chem. C 2015, 119, 7491–7498. [Google Scholar] [CrossRef]
- Chistyakov, A.A.; Zvaigzne, M.A.; Nikitenko, V.R.; Tameev, A.R.; Martynov, I.L.; Prezhdo, O.V. Optoelectronic Properties of Semiconductor Quantum Dot Solids for Photovoltaic Applications. J. Phys. Chem. Lett. 2017, 8, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Engel, G.S.; Kais, S. Double-excitation manifold’s effect on exciton transfer dynamics and the efficiency of coherent light harvesting. Phys. Chem. Chem. Phys. 2018, 20, 30032–30040. [Google Scholar] [CrossRef]
- Yan, T.-M.; Fresch, B.; Levine, R.D.; Remacle, F. Information processing in parallel through directionally resolved molecular polarization components in coherent multidimensional spectroscopy. J. Chem. Phys. 2015, 143, 064106. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gattuso, H.; Fresch, B.; Levine, R.D.; Remacle, F. Coherent Exciton Dynamics in Ensembles of Size-Dispersed CdSe Quantum Dot Dimers Probed via Ultrafast Spectroscopy: A Quantum Computational Study. Appl. Sci. 2020, 10, 1328. https://doi.org/10.3390/app10041328
Gattuso H, Fresch B, Levine RD, Remacle F. Coherent Exciton Dynamics in Ensembles of Size-Dispersed CdSe Quantum Dot Dimers Probed via Ultrafast Spectroscopy: A Quantum Computational Study. Applied Sciences. 2020; 10(4):1328. https://doi.org/10.3390/app10041328
Chicago/Turabian StyleGattuso, Hugo, Barbara Fresch, Raphael D. Levine, and Françoise Remacle. 2020. "Coherent Exciton Dynamics in Ensembles of Size-Dispersed CdSe Quantum Dot Dimers Probed via Ultrafast Spectroscopy: A Quantum Computational Study" Applied Sciences 10, no. 4: 1328. https://doi.org/10.3390/app10041328
APA StyleGattuso, H., Fresch, B., Levine, R. D., & Remacle, F. (2020). Coherent Exciton Dynamics in Ensembles of Size-Dispersed CdSe Quantum Dot Dimers Probed via Ultrafast Spectroscopy: A Quantum Computational Study. Applied Sciences, 10(4), 1328. https://doi.org/10.3390/app10041328