Mechanistic Studies of Hydrogen Evolution Reaction on Donor-Acceptor Conjugated Polymer Photocatalysts



 ,
, 
Abstract
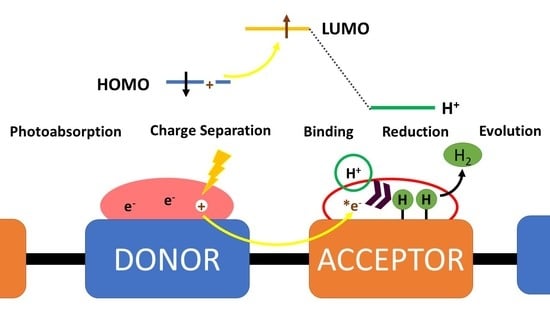
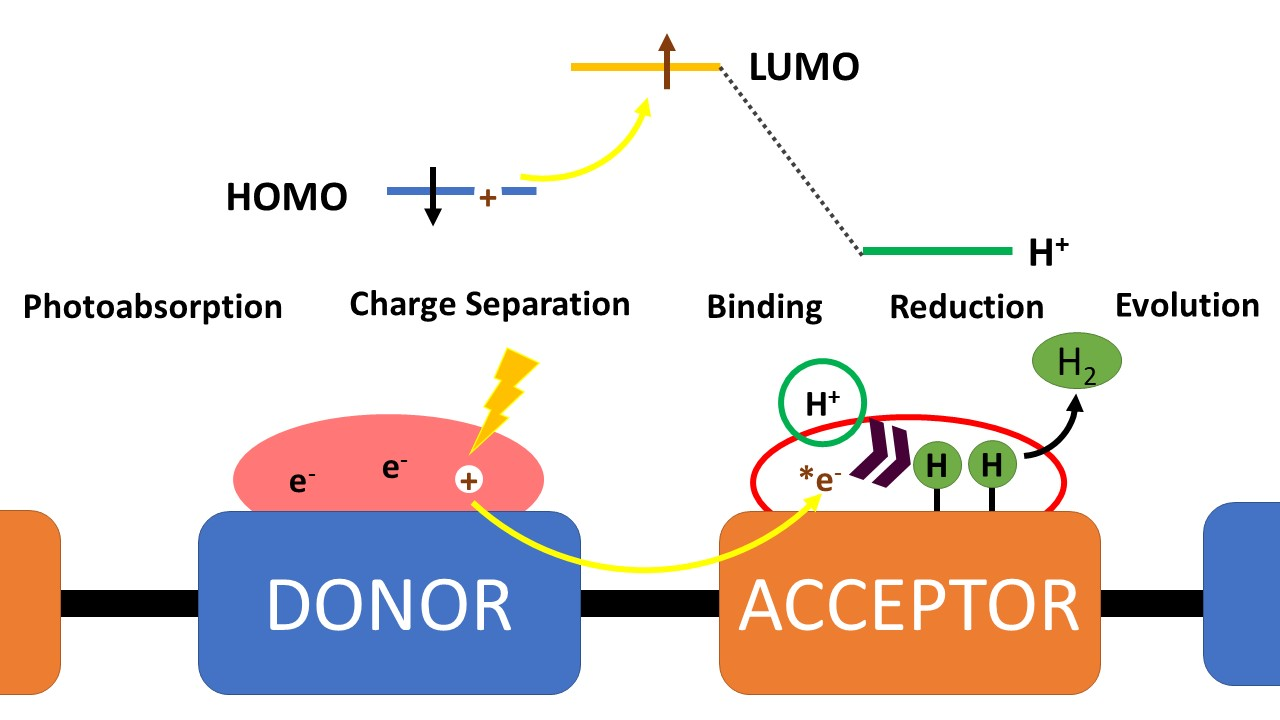
1. Introduction
2. Materials and Methods
2.1. General Method
2.1.1. Synthesis of 3,6-dinitro-9H-carbazole
2.1.2. Synthesis of 3,6-dinitro-9-(4-nitrophenyl)-9H-carbazole
2.1.3. Synthesis of 9-(4-aminophenyl)-carbazole-3,6-diamine
2.1.4. Synthesis of tris(2-fluoro-4-nitrophenyl)amine
2.1.5. Synthesis of tris(2-fluoro-4-aminophenyl)amine
2.1.6. Synthesis of 4,4′-(5-oxido-5-phenylbenzo[b]phosphindole-3,7-diyl)dibenzaldehyde
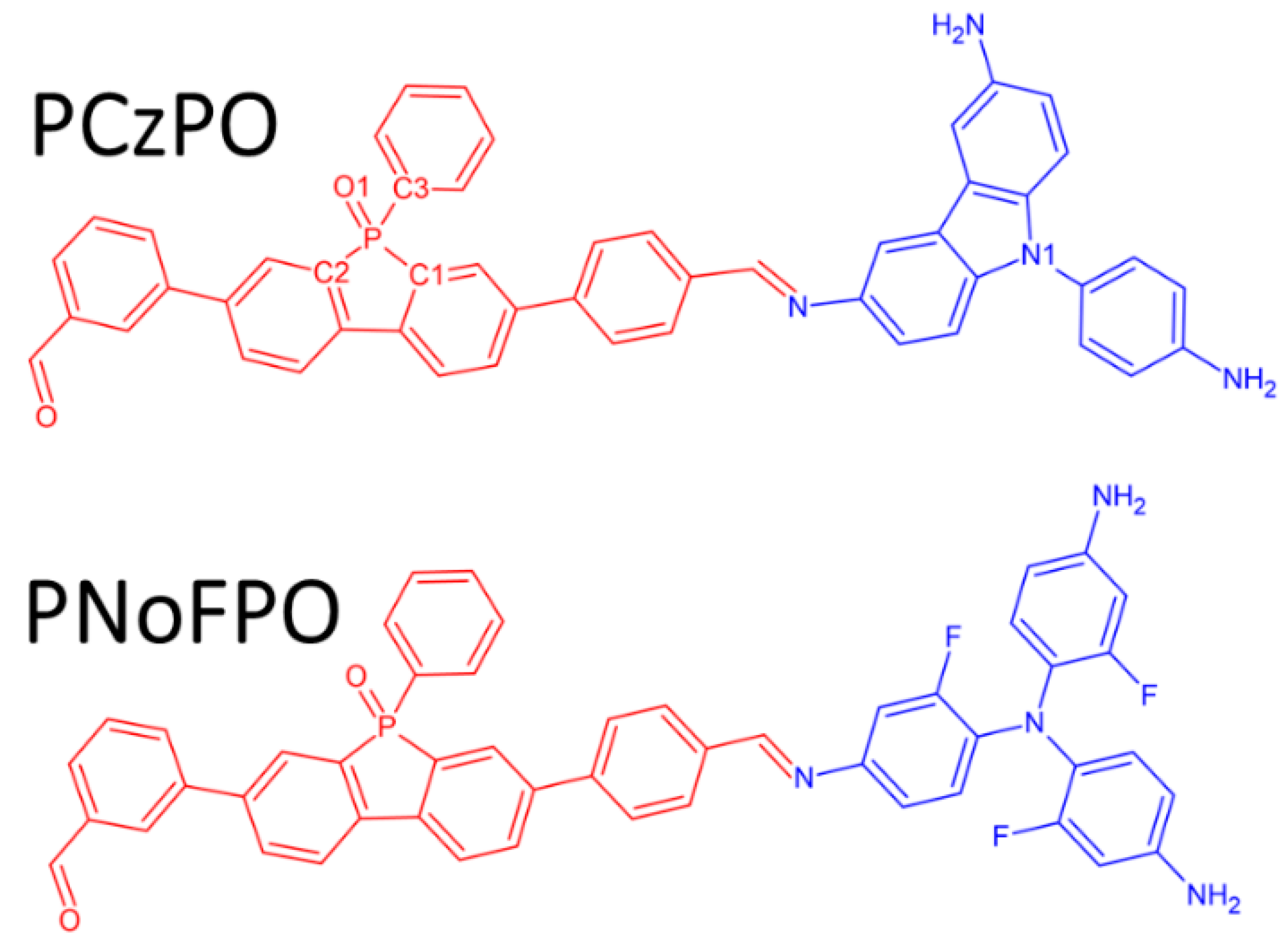
2.1.7. Synthesis of PCzPO
2.1.8. Synthesis of PNoFPO
2.2. Hydrogen Evolution Experiments
2.3. Computational Details
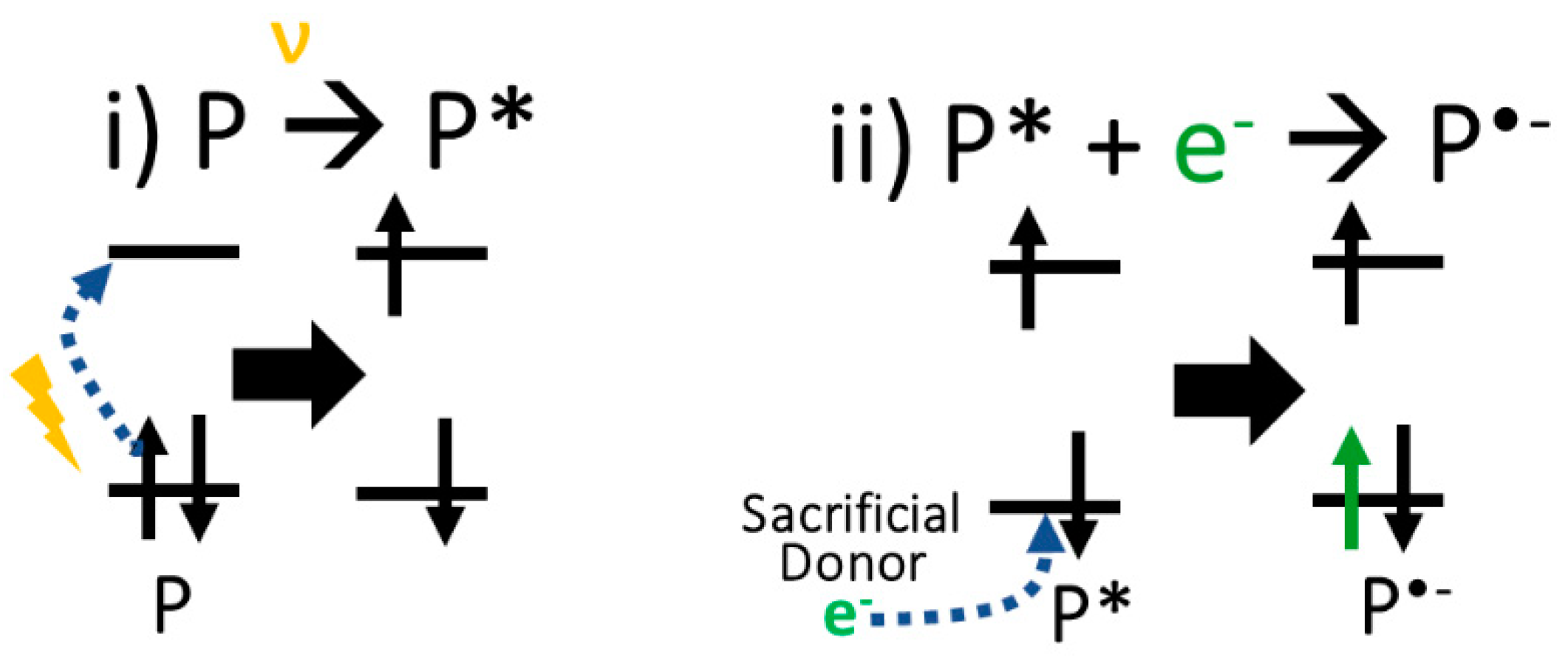
3. Results and Discussions
3.1. Experimental Characterisation and HER Performance
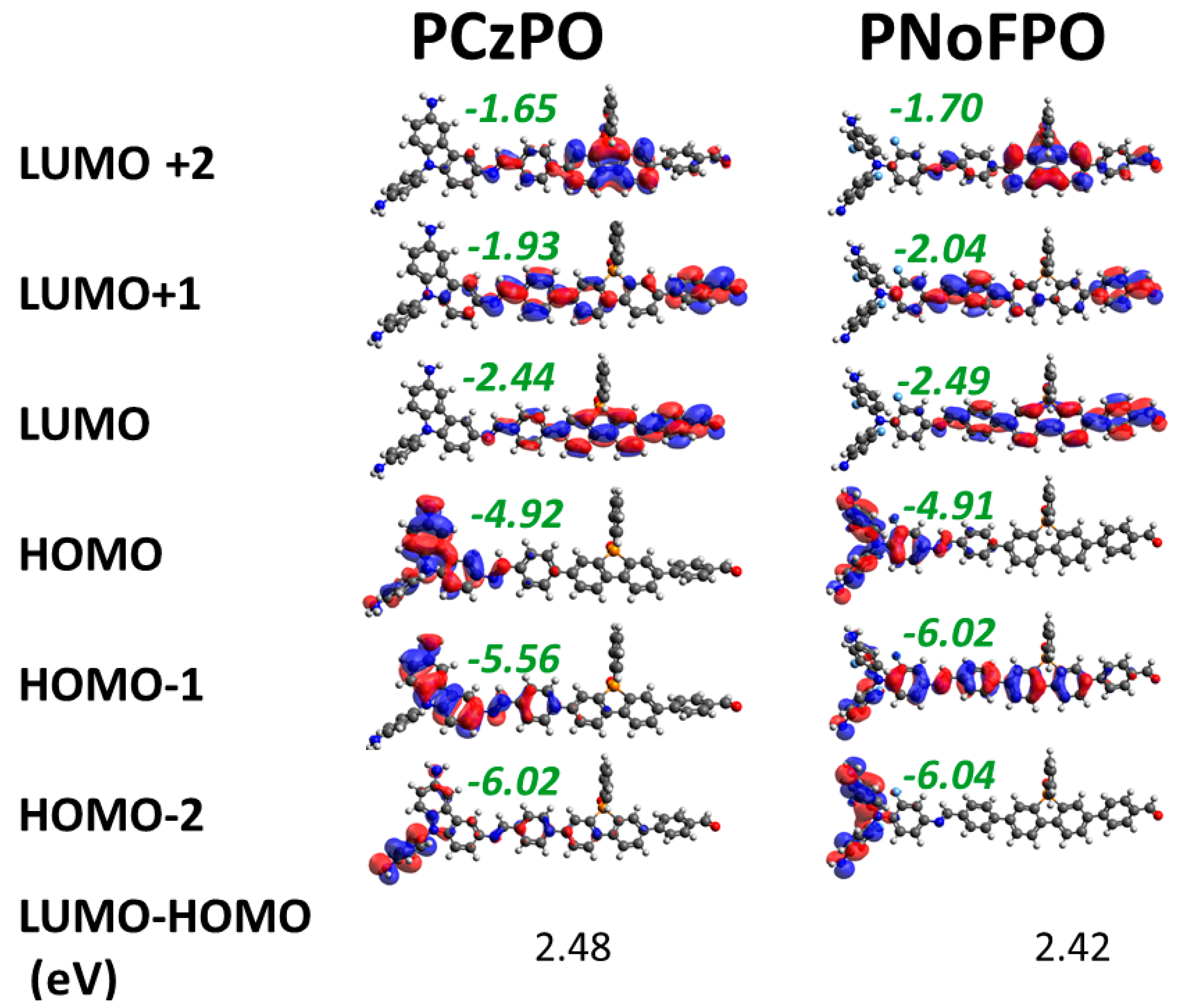
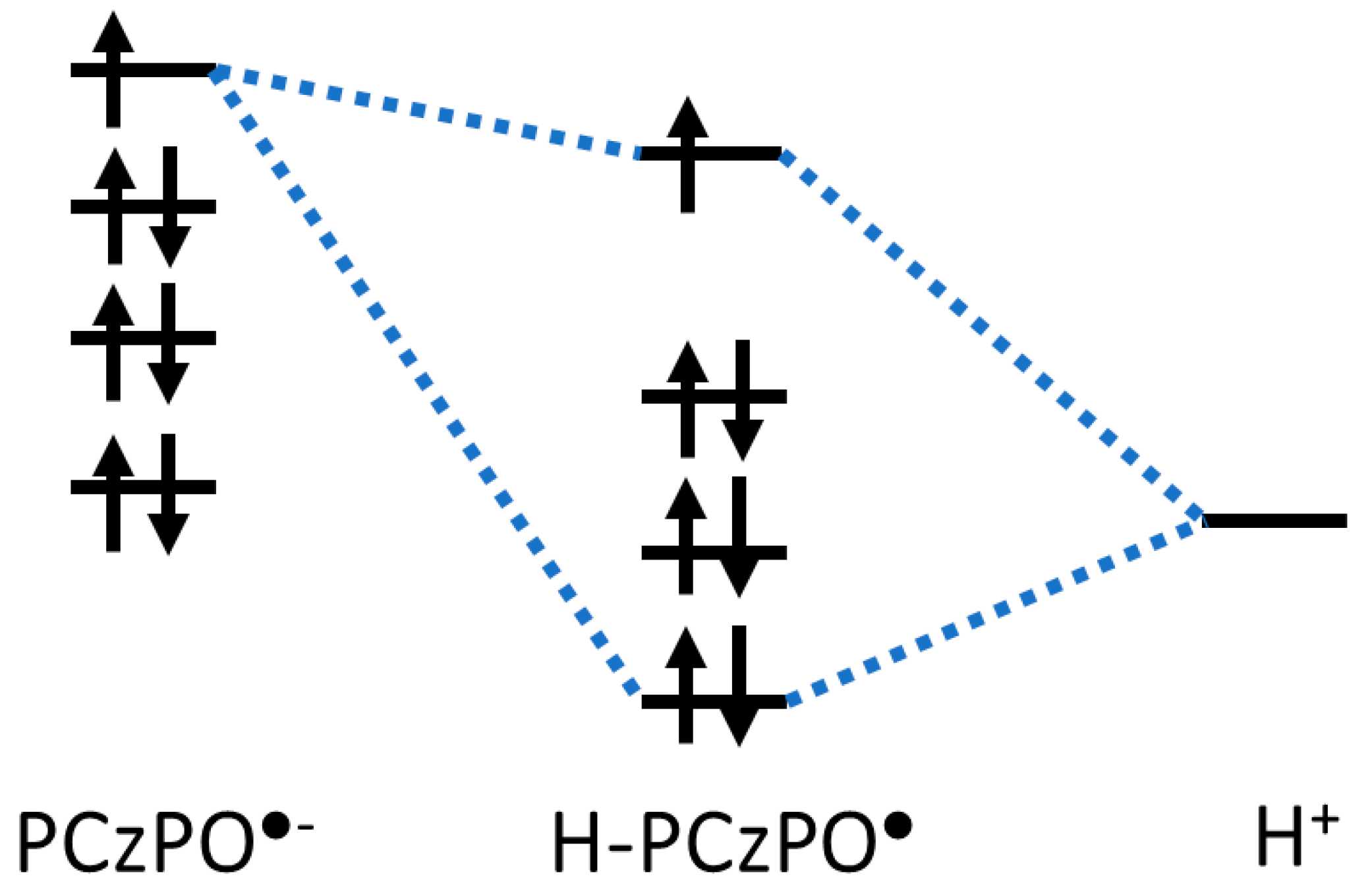
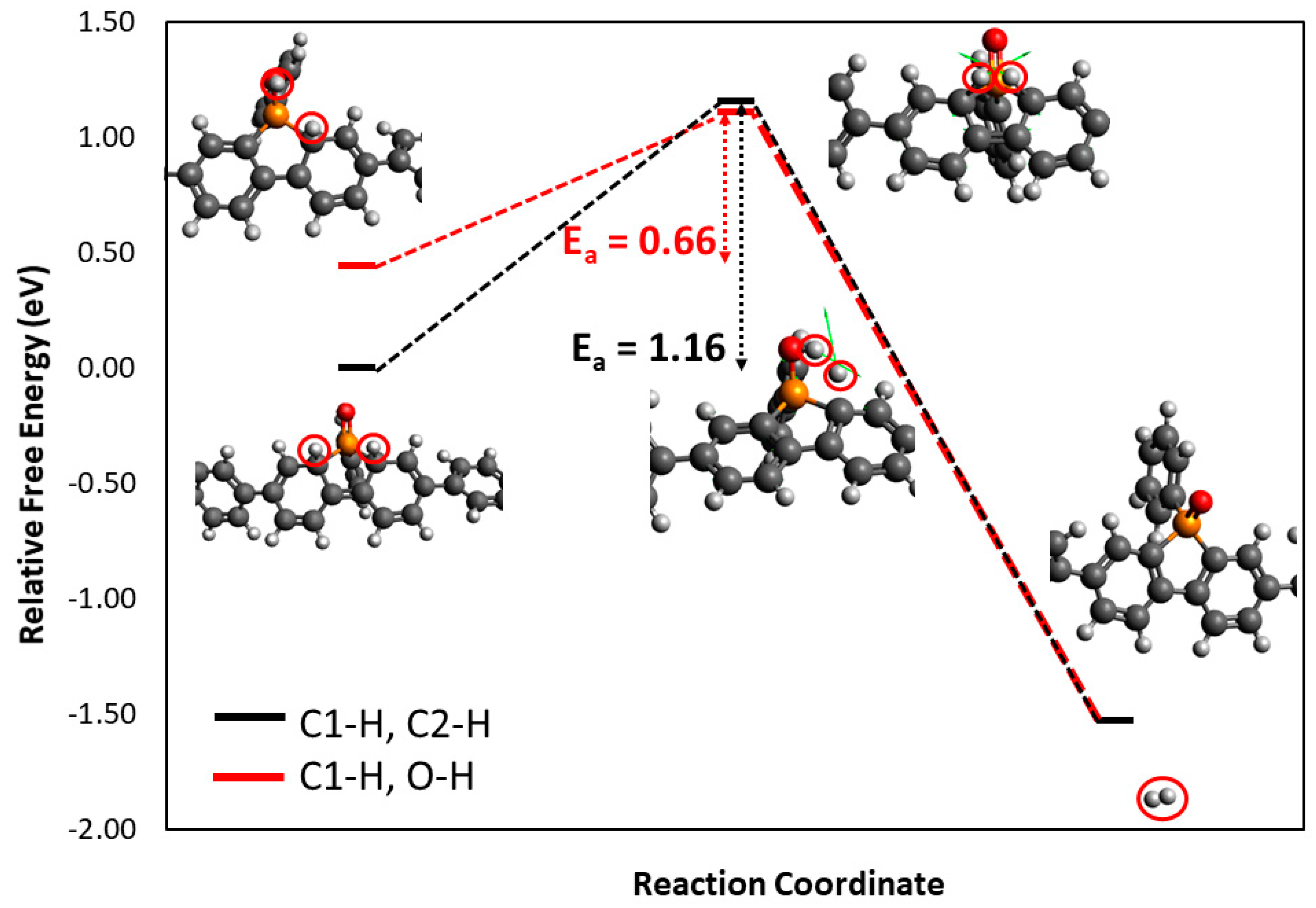
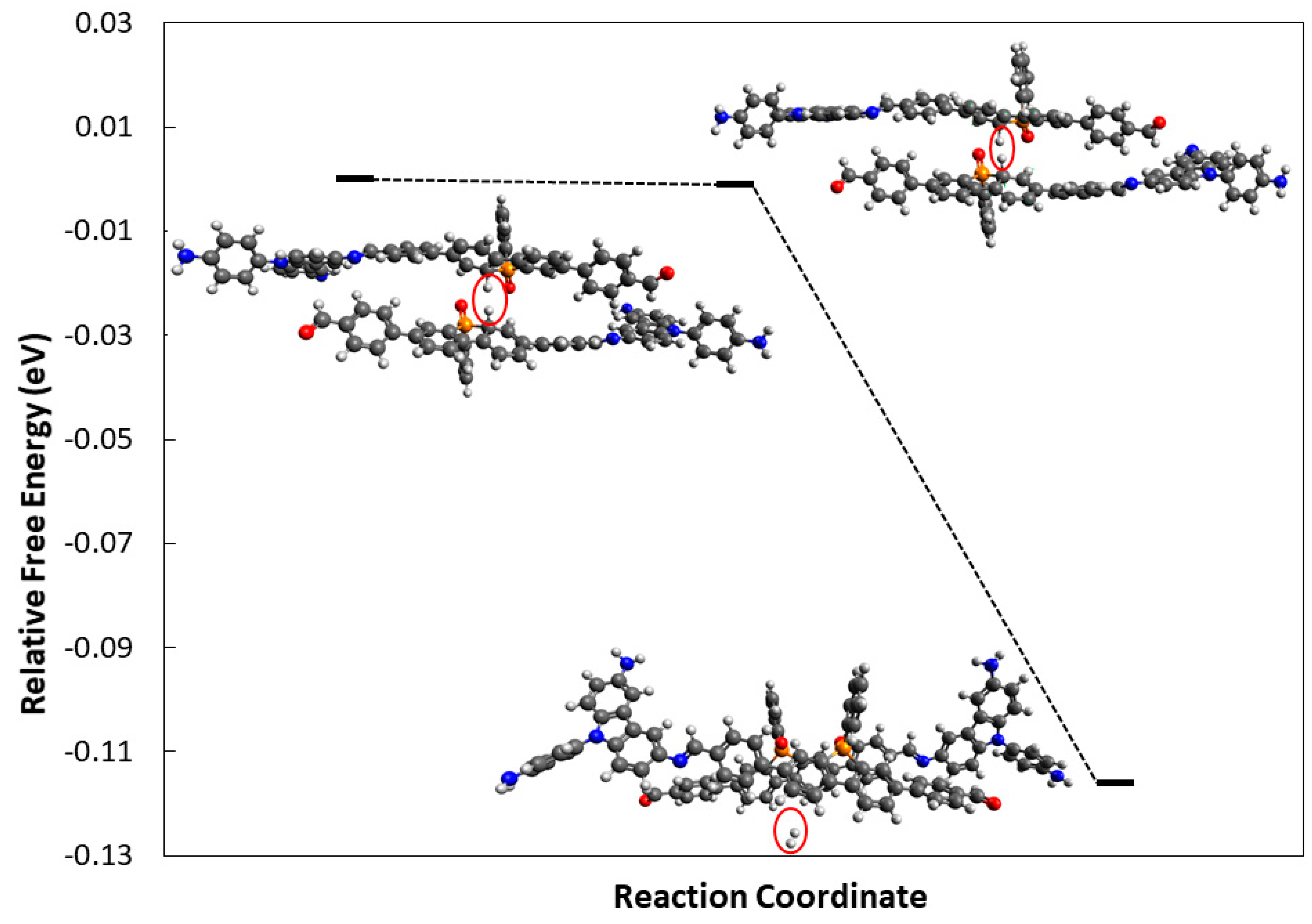
3.2. Density Functional Theory Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tachibana, Y.; Vayssieres, L.; Durrant, J.R. Artificial photosynthesis for solar water-splitting. Nat. Photonics 2012, 6, 511–518. [Google Scholar] [CrossRef]
- Jain, A.; Shin, Y.; Persson, K.A. Computational predictions of energy materials using density functional theory. Nat. Rev. Mater. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Jayakumar, J.; Chou, H. Recent advances in visible-light-driven hydrogen evolution from water using polymer photocatalysts. ChemCatChem 2020, 12, 689–704. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.L.; Lau, T.K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Zhou, N.; Dudnik, A.S.; Li, T.I.N.G.; Manley, E.F.; Aldrich, T.J.; Guo, P.; Liao, H.C.; Chen, Z.; Chen, L.X.; Chang, R.P.H.; et al. All-polymer solar cell performance optimized via systematic molecular weight tuning of both donor and acceptor polymers. J. Am. Chem. Soc. 2016, 138, 1240–1251. [Google Scholar] [CrossRef]
- Benten, H.; Mori, D.; Ohkita, H.; Ito, S. Recent research progress of polymer donor/polymer acceptor blend solar cells. J. Mater. Chem. A 2016, 4, 5340–5365. [Google Scholar] [CrossRef]
- Tseng, P.J.; Chang, C.L.; Chan, Y.H.; Ting, L.Y.; Chen, P.Y.; Liao, C.H.; Tsai, M.L.; Chou, H.H. Design and synthesis of cycloplatinated polymer dots as photocatalysts for visible-light-driven hydrogen evolution. ACS Catal. 2018, 8, 7766–7772. [Google Scholar] [CrossRef]
- EL-Mahdy, A.F.M.; Elewa, A.M.; Huang, S.; Chou, H.; Kuo, S. Dual-function fluorescent covalent organic frameworks: HCl sensing and photocatalytic H2 evolution from water. Adv. Opt. Mater. 2020, 2000641. [Google Scholar] [CrossRef]
- Wang, W.H.; Ting, L.Y.; Jayakumar, J.; Chang, C.L.; Lin, W.C.; Chung, C.C.; Elsayed, M.H.; Lu, C.Y.; Elewa, A.; Chou, H.H.; et al. Design and synthesis of phenylphosphine oxide-based polymer photocatalysts for highly efficient visible-light-driven hydrogen evolution. Sustain. Energy Fuels 2020, 4, 5264–5270. [Google Scholar] [CrossRef]
- Pati, P.B.; Damas, G.; Tian, L.; Fernandes, D.L.A.; Zhang, L.; Pehlivan, I.B.; Edvinsson, T.; Araujo, C.M.; Tian, H. An experimental and theoretical study of an efficient polymer nano-photocatalyst for hydrogen evolution. Energy Environ. Sci. 2017, 10, 1372–1376. [Google Scholar] [CrossRef]
- Banerjee, T.; Gottschling, K.; Savasci, G.; Ochsenfeld, C.; Lotsch, B.V. H2 evolution with covalent organic framework photocatalysts. ACS Energy Lett. 2018, 3, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Hug, S.; Mesch, M.B.; Oh, H.; Popp, N.; Hirscher, M.; Senker, J.; Lotsch, B.V. A fluorene based covalent triazine framework with high CO2 and H2 capture and storage capacities. J. Mater. Chem. A 2014, 2, 5928–5936. [Google Scholar] [CrossRef]
- Lee, W.Y.; Cheng, K.F.; Wang, T.F.; Chueh, C.C.; Chen, W.C.; Tuan, C.S.; Lin, J.L. Effects of acceptors on the electronic and optoelectronic properties of fluorene-based donor–Acceptor–donor copolymers. Macromol. Chem. Phys. 2007, 208, 1919–1927. [Google Scholar] [CrossRef]
- Guo, L.; Niu, Y.; Xu, H.; Li, Q.; Razzaque, S.; Huang, Q.; Jin, S.; Tan, B. Engineering heteroatoms with atomic precision in donor-acceptor covalent triazine frameworks to boost photocatalytic hydrogen production. J. Mater. Chem. A 2018, 6, 19775–19781. [Google Scholar] [CrossRef]
- Bibi, S.; Bilal, S.; Ali Shah, A.U.H.; Ullah, H. Systematic analysis of poly(o-aminophenol) humidity sensors. ACS Omega 2017, 2, 6380–6390. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Bibi, S.; Tahir, A.A.; Mallick, T.K. Donor-acceptor polymer for the design of all-solid-state dye-sensitized solar cells. J. Alloys Compd. 2017, 696, 914–922. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, X.; Rao, L.; Wang, P.; Huang, D.; Ding, X.; Zhang, X.; Wang, S.; Chen, H.; Zhu, Y.; et al. Conjugated polymers with sequential fluorination for enhanced photocatalytic H2 evolution via proton-coupled electron transfer. ACS Energy Lett. 2018, 3, 2544–2549. [Google Scholar] [CrossRef]
- Bai, Y.; Woods, D.J.; Wilbraham, L.; Aitchison, C.M.; Zwijnenburg, M.A.; Sprick, R.S.; Cooper, A.I. Hydrogen evolution from water using heteroatom substituted fluorene conjugated co-polymers. J. Mater. Chem. A 2020, 8, 8700–8705. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of hartree–fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Franco, F.C.; Padama, A.A.B. DFT and TD-DFT study on the structural and optoelectronic characteristics of chemically modified donor-acceptor conjugated oligomers for organic polymer solar cells. Polymer 2016, 97, 55–62. [Google Scholar] [CrossRef]
- Aicha Youssef, A.; Mohamed Bouzzine, S.; Mohyi Eddine Fahim, Z.; Sıdır, İ.; Hamidi, M.; Bouachrine, M. Designing donor-acceptor thienopyrazine derivatives for more efficient organic photovoltaic solar cell: A DFT study. Phys. B Condens. Matter. 2019, 560, 111–125. [Google Scholar] [CrossRef]
- Lan, Z.A.; Ren, W.; Chen, X.; Zhang, Y.; Wang, X. Conjugated donor-acceptor polymer photocatalysts with electron-output “tentacles” for efficient hydrogen evolution. Appl. Catal. B Environ. 2019, 245, 596–603. [Google Scholar] [CrossRef]
- Patra, B.C.; Khilari, S.; Manna, R.N.; Mondal, S.; Pradhan, D.; Pradhan, A.; Bhaumik, A. A metal-free covalent organic polymer for electrocatalytic hydrogen evolution. ACS Catal. 2017, 7, 6120–6127. [Google Scholar] [CrossRef]
- Chen, B.S.; Chen, D.Y.; Chen, C.L.; Hsu, C.W.; Hsu, H.C.; Wu, K.L.; Liu, S.H.; Chou, P.T.; Chi, Y. Donor-acceptor dyes with fluorine substituted phenylene spacer for dye-sensitized solar cells. J. Mater. Chem. 2011, 21, 1937–1945. [Google Scholar] [CrossRef]
- Afroz, M.A.; Sonigara, K.K.; Bhim Raju, T.; Soni, S.S.; Iyer, P.K. Effect of fluorine substitution and position on phenylene spacer in carbazole based organic sensitizers for dye sensitized solar cells. Phys. Chem. Chem. Phys. 2017, 19, 28579–28587. [Google Scholar] [CrossRef]
- Ting, L.Y.; Jayakumar, J.; Chang, C.L.; Lin, W.C.; Elsayed, M.H.; Chou, H.H. Effect of controlling the number of fused rings on polymer photocatalysts for visible-light-driven hydrogen evolution. J. Mater. Chem. A 2019, 7, 22924–22929. [Google Scholar] [CrossRef]
- Bai, Y.; Wilbraham, L.; Slater, B.J.; Zwijnenburg, M.A.; Sprick, R.S.; Cooper, A.I. Accelerated discovery of organic polymer photocatalysts for hydrogen evolution from water through the integration of experiment and theory. J. Am. Chem. Soc. 2019, 141, 9063–9071. [Google Scholar] [CrossRef]
- Bredas, J.L. Mind the gap! Mater. Horiz. 2014, 1, 17–19. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Absorption Peaks [nm] | HOMO/LUMO [eV] a,b | Optical Gap [eV] c | HER Rate [µmol h−1 g−1] d |
|---|---|---|---|---|
| PCzPO | 334, 357 | −5.59/−2.85 | 2.74 | 34.23 |
| PNoFPO | 342, 368 | −5.87/−2.84 | 3.03 | 7.34 |
| Polymer Catalyst | Proton Binding Free Energy (eV) | |
|---|---|---|
| P + H+ → PH+ | P•- + H+ → PH | |
| PCzPO | 5.93 | 2.96 |
| PNoFPO | 5.95 | 3.02 |
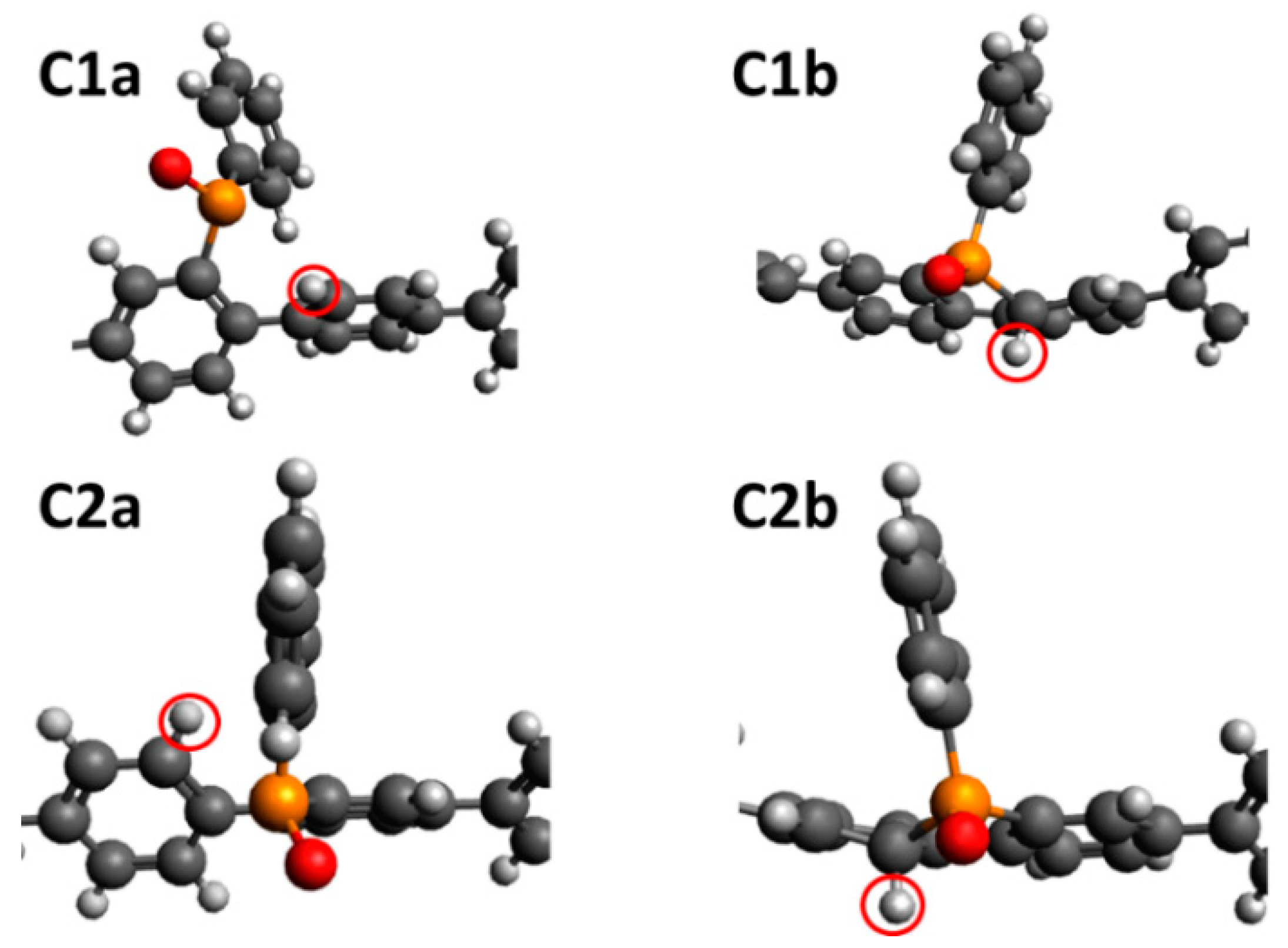
| Site | Configuration | Mulliken Charge | ΔGH+ (eV) |
|---|---|---|---|
| C1 | a | −0.417 | 2.83 |
| b | 2.98 | ||
| C2 | a | −0.412 | 2.90 |
| b | 2.99 | ||
| C3 | −0.382 | 3.07 | |
| O | −0.593 | 2.97 | |
| N1 | −0.593 | 4.37 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, Y.I.A.; Ting, L.-Y.; Tu, X.; Chen, H.-Y.T.; Chou, H.-H.; Coluccini, C. Mechanistic Studies of Hydrogen Evolution Reaction on Donor-Acceptor Conjugated Polymer Photocatalysts. Appl. Sci. 2020, 10, 7017. https://doi.org/10.3390/app10207017
Reyes YIA, Ting L-Y, Tu X, Chen H-YT, Chou H-H, Coluccini C. Mechanistic Studies of Hydrogen Evolution Reaction on Donor-Acceptor Conjugated Polymer Photocatalysts. Applied Sciences. 2020; 10(20):7017. https://doi.org/10.3390/app10207017
Chicago/Turabian StyleReyes, Yves Ira A., Li-Yu Ting, Xin Tu, Hsin-Yi Tiffany Chen, Ho-Hsiu Chou, and Carmine Coluccini. 2020. "Mechanistic Studies of Hydrogen Evolution Reaction on Donor-Acceptor Conjugated Polymer Photocatalysts" Applied Sciences 10, no. 20: 7017. https://doi.org/10.3390/app10207017
APA StyleReyes, Y. I. A., Ting, L.-Y., Tu, X., Chen, H.-Y. T., Chou, H.-H., & Coluccini, C. (2020). Mechanistic Studies of Hydrogen Evolution Reaction on Donor-Acceptor Conjugated Polymer Photocatalysts. Applied Sciences, 10(20), 7017. https://doi.org/10.3390/app10207017