Reaction of Ion Exchange Resins with Fenton’s Reagent

Abstract
:1. Introduction
2. Materials and Methods
2.1. Total Organic Carbon Concentration (TOC)
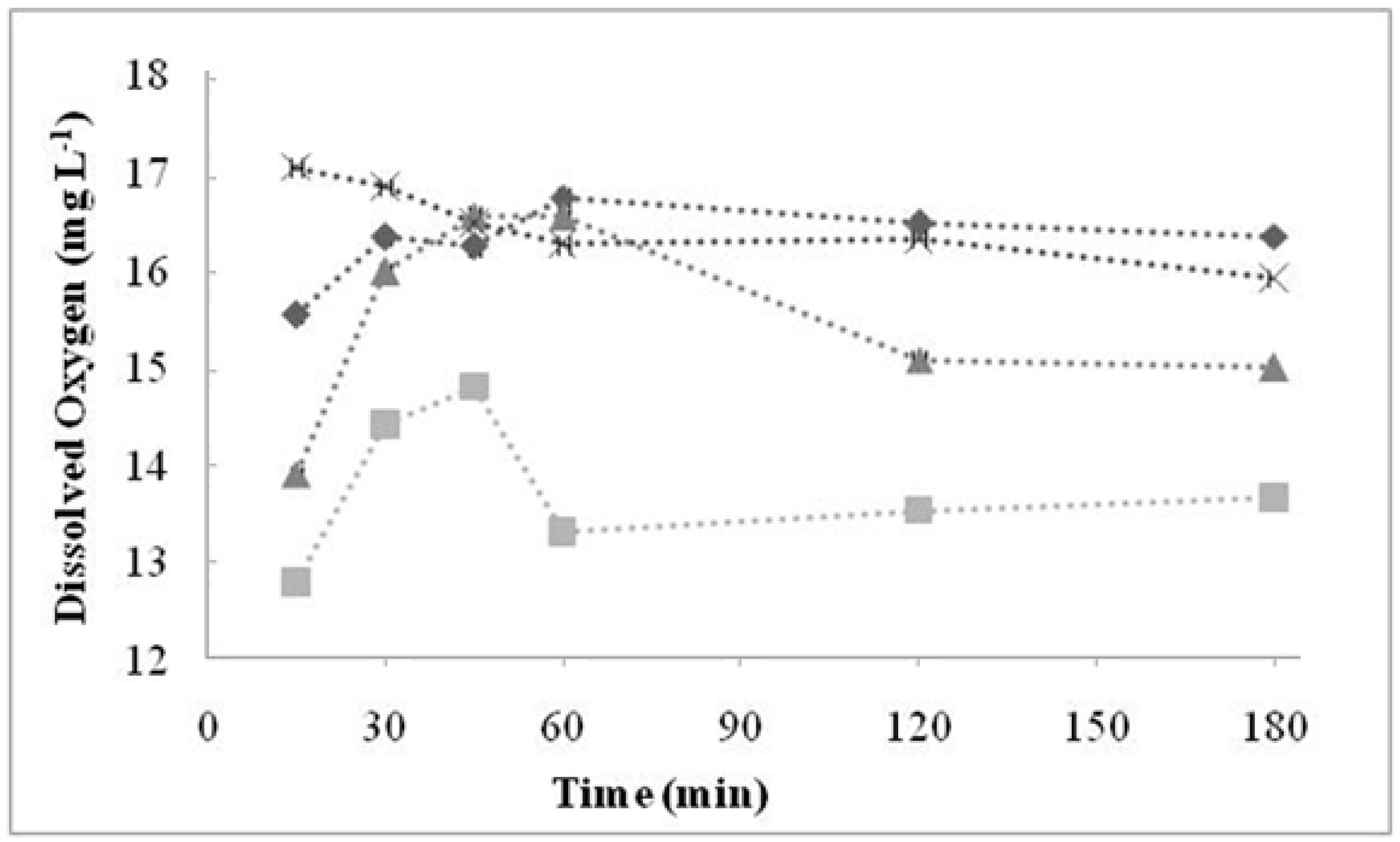
2.2. Dissolved Oxygen (DO)
2.3. Fourier Transform Infrared Spectrometry (FTIR)
2.4. Residual Resin Mass
2.5. Cementation
3. Results and Discussion
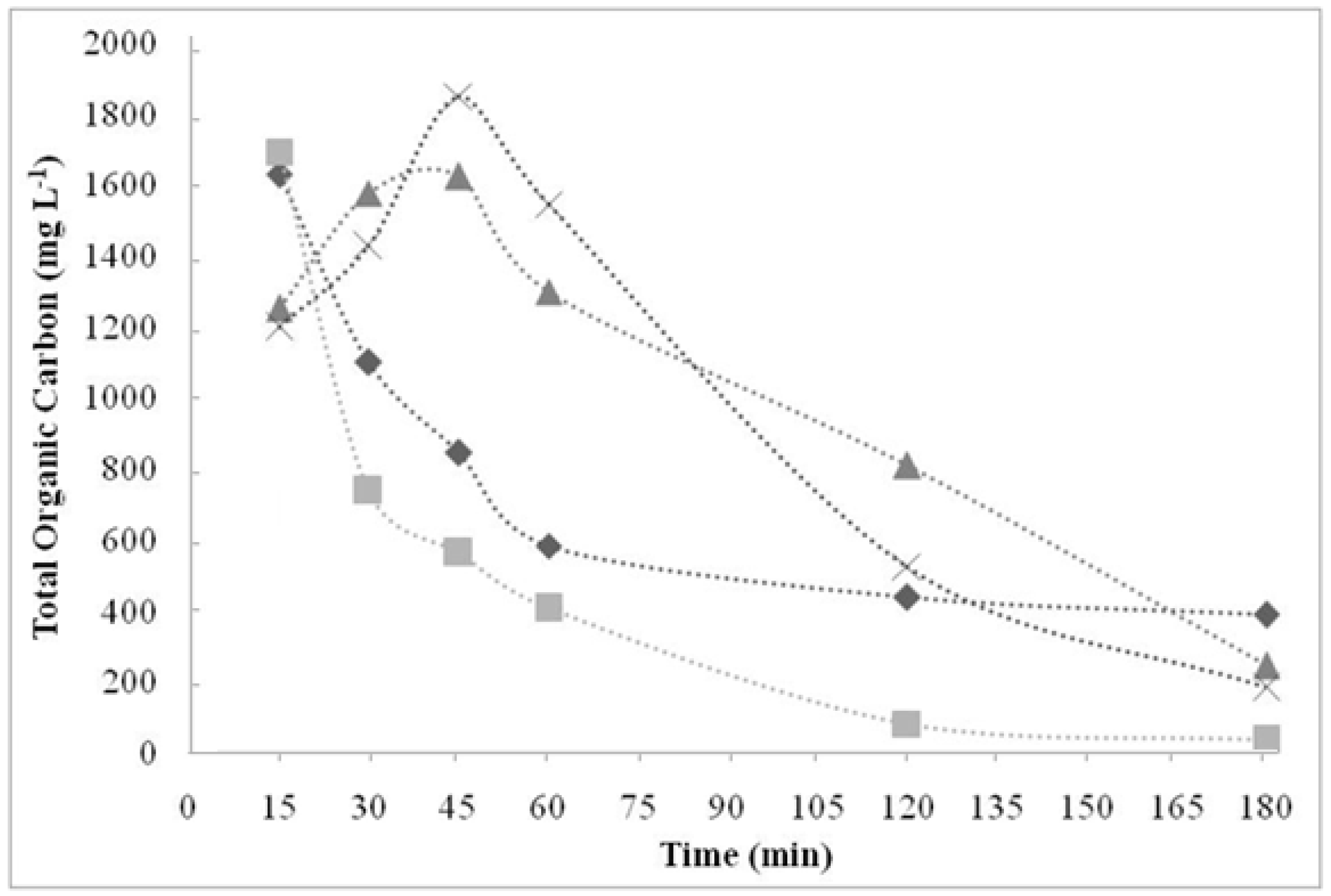
3.1. Effects of Temperature and Catalyst Concentration
3.2. Immobilization with Portland Cement
4. Conclusions
- The catalyst concentration interferes in oxidation reaction. Concentrations of 50 and 100 mmol L−1 were more effective than those of 25 and 150 mmol L−1. However, the 50 mmol L−1 was the most efficient, since less catalyst is required compared to that of 100 mmol L−1 to treat 10 g of resin.
- It was possible to degrade resins efficiently without external heating. The temperature of 60 °C was the most adequate.
- As predicted, TOC was efficient as a reaction parameter to determine resin degradation rates.
- The suspension/cement ratio of 0.28 was suitable to produce an immobilized product, which comply with the limits established by Brazilian regulation.
- The evaluated degradation processes and the evaporation allowed the incorporation of a mass three-fold higher when compared with direct immobilization.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, J.; Wang, J. Advances in cement solidification technology for waste radioactive ion exchange resins: A review. J. Hazard. Mater. 2006, 135, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Matilainen, A.; Sillanpää, M. Removal of natural organic matter from drinking water by advanced oxidation processes. Chemosphere 2010, 80, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.A. Destruction of Ion-Exchange Resin in Waste from the HFIR, T1, and T2 Tanks Using Fenton’s Reagent. OSTI GOA 2002. [Google Scholar] [CrossRef]
- Zahorodna, M.; Bogoczek, R.; Oliveros, E.; Braun, A.M. Application of the Fenton process to the dissolution and mineralization of ion exchange resins. Chem. Eng. J. 2007, 129, 200–206. [Google Scholar] [CrossRef]
- Araujo, F.V.F. Heterogeneous Fenton process using the mineral hematite for the discolouration of a reactive dye solution. Braz. J. Chem. Eng. 2011, 8, 605–616. [Google Scholar] [CrossRef]
- Jian, X.; Wu, T.; Yun, G. A Study of Wet Catalytic Oxidation of Radioactive Spent Ion Exchange Resin by Hydrogen Peroxide. Nucl. Saf. 1996, 37, 149–157. [Google Scholar]
- Nogueira, R.F.P.; Trovó, A.G.; Silva, M.R.A.; Villa, R.D.; Oliveira, M.C. Fundamentals and environmental applications of Fenton and Photo-Fenton processes. Quim. Nova 2007, 30, 400–408. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (–OH/–O) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Geng, Z.; Wu, J.; Yun, G.; Wu, T. A study on free radical oxidation of spent radioactive ion-exchange resins. INIS 1993, 14, 66–76. [Google Scholar]
- Park, S.; Yoon, T. The effects of iron species and mineral particles on advanced oxidation processes for the removal of humic acids. Desalination 2007, 208, 181–191. [Google Scholar] [CrossRef]
- Moncayo-Lasso, A.; Pulgarin, C.; Benítez, N. Degradation of DBPs’ precursors in river water before and after slow sand filtration by photo-Fenton process at pH 5 in solar CPC reactor. Water Res. 2008, 42, 4125–4132. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wan, Z. Treatment and disposal of spent radioactive ion-exchange resins produced in the nuclear industry. Prog. Nucl. Energy 2015, 78, 47–55. [Google Scholar] [CrossRef]
- Wan, Z.; Xu, L.; Wang, J. Treatment of spent radioactive anionic exchange resins using Fenton-like oxidation process. Chem. Eng. J. 2016, 284, 733–740. [Google Scholar] [CrossRef]
- Zagorodni, A.; Kotova, D.L.; Selemenev, V.F. Infrared spectroscopy of ion exchange resins: Chemical deterioration of the resins. React. Funct. Polym. 2002, 53, 157–171. [Google Scholar] [CrossRef]
- NBR 11581, Cimento Portland-Determinação dos Tempos de Pega. Available online: http://professor.pucgoias.edu.br/SiteDocente/admin/arquivosUpload/17827/material/NBR%20NM%2065%20-.pdf (accessed on 13 November 2018).
- Rosenfeldt, E.J.; Linden, K.G. The ROH, UV concept to characterize and the model UV/H2O2 process in natural waters. Environ. Sci. Technol. 2007, 41, 2548–2553. [Google Scholar] [CrossRef]
- Juanes, L.S.; Ballesteros, M.M.; Ortega, E.; Cabrera, A.; Román, L.M.; Casas, J.L.; Sánchez, J.A. Controlling dissolved oxygen concentration as a tool for economical efficiency improvement in a solar photo-Fenton treatment plant. In Proceedings of the 8th IWA International Conference on Water Reclamation & Reuse, Barcelona, Spain, 22–29 September 2011. [Google Scholar]
- Brazilian Nuclear Energy Commission. CNEN-NN-6.09: Regulation on Acceptance Criteria for Disposal of Radioactive Waste of Low and Medium Levels of Radiation; Brazilian Nuclear Energy Commission: Rio de Janeiro, Spain, 2002. (In Portuguese)
- Ye, Y.C.; Wang, R. The research of strength and formula research of the cementation of radioactive waste ion exchange resins. Atom. Energy Sci. Technol. 1983, 264–268. [Google Scholar]
- Zhou, Y.Z.; Ye, Y.C.; Yun, G.C.; Zhang, M. Study on solidification of spent ion exchange resin using ASC cement. Radiat. Prot. 2002, 22, 225–230. [Google Scholar]
- Ollivier, J.P.; École, F.D.B.A. La Durabilité Des Bétons (avec CD-ROM): Bases Scientifiques Pour La Formulation De Bétons Durables Dans Leur Environnement; de l’école nationale des Ponts et Chaussées Presses: Paris, France, 2008. [Google Scholar]
- Isiki, V.L.K. Evaluation of Portland Cement as an Immobilization Matrix of Activated Carbon from IPEN-CNEN/SP Research Reactor. Bachelor’s Thesis, Faculdades Oswaldo Cruz, São Paulo, Brazil, 2003. [Google Scholar]
- Quennoz, A.; Galucci, E.; Scrivener, K.L. Calcium silicate-calcium aluminate interactions and their influence on cement early hydration. In Proceedings of the XIII ICCC INTERNATIONAL Congress on the Chemistry of Cement, Madrid, Spain, 3–8 July 2011. [Google Scholar]
 ) 25; (
) 25; (  ) 50; (
) 50; (  ) 100; (
) 100; ( 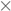 ) 150.
) 25; ( ) 50; ( ) 100; ( ) 150.
) 150.
) 25; ( ) 50; ( ) 100; ( ) 150.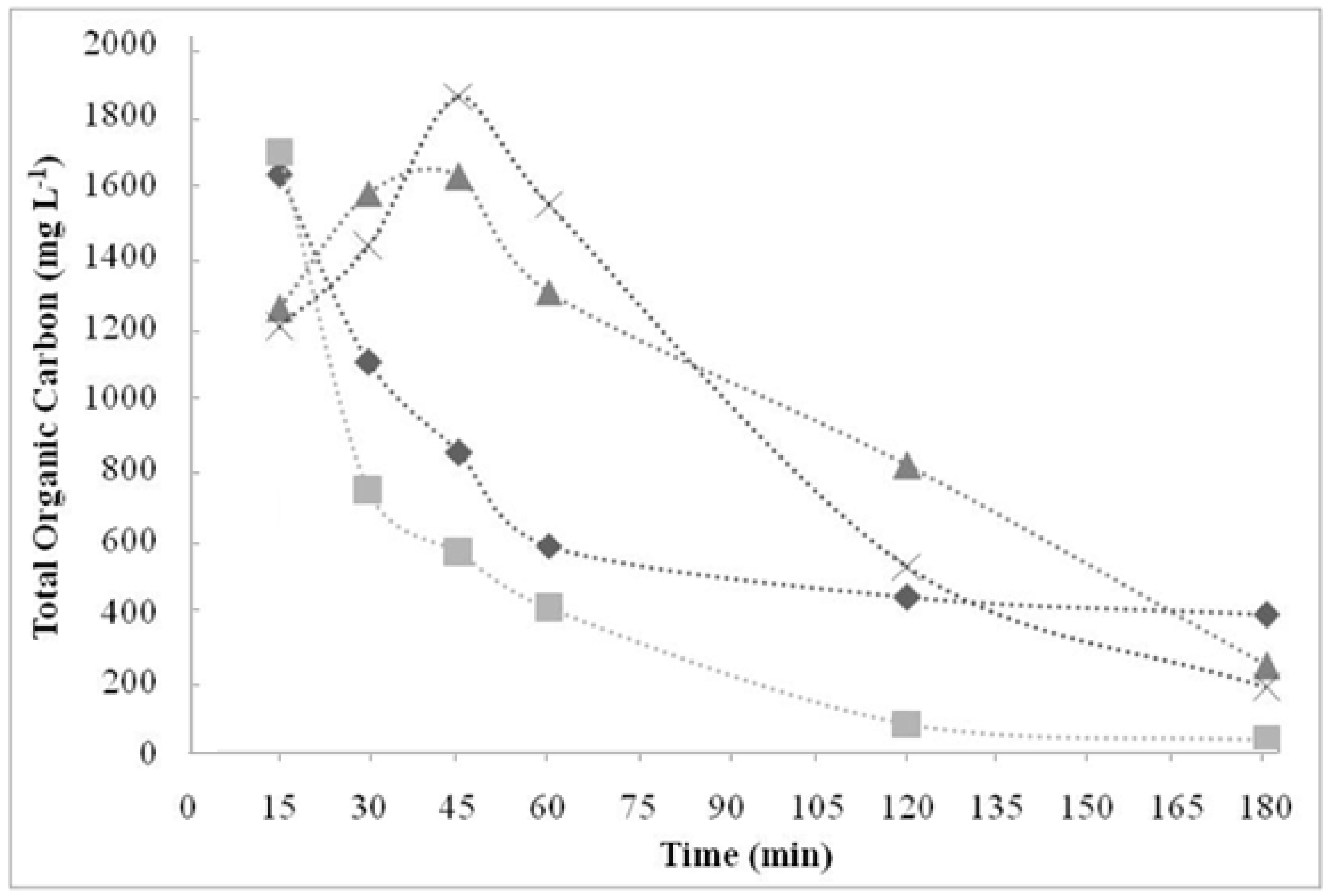 ) 25; ( ) 50; ( ) 100; ( ) 150.
) 25; ( ) 50; ( ) 100; ( ) 150.
) 25; ( ) 50; ( ) 100; ( ) 150.
) 25; ( ) 50; ( ) 100; ( ) 150.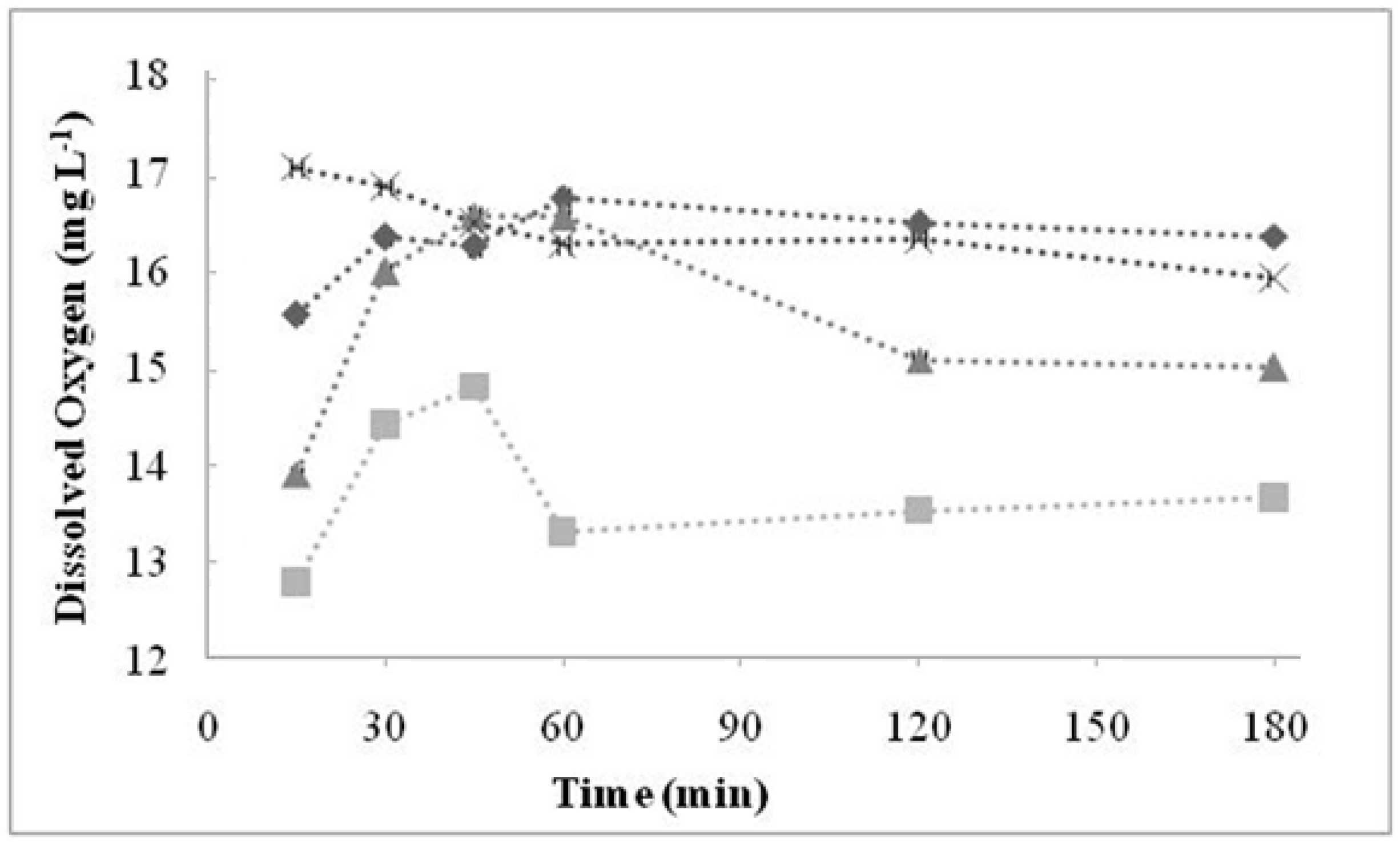

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FeSO4·7H2O Concentration (mmol L−1) | H2O2 Volume (mL) |
|---|---|
| 25 | 460 |
| 50 | 335 |
| 100 | 350 |
| 150 | 340 |
| FeSO4·7H2O Concentration (mmol L−1) | Dry Resin Mass (g) | Temperature (°C) α | H2O2 25% Volume (mL) α | Residual Resin Mass (g) α | Average Degradation (%) |
|---|---|---|---|---|---|
| 25 | 5.6 | 41 ± 1 | 480 ± 20 | 1.56 ± 0.03 | 76.14 |
| 50 | 5.6 | 60 ± 6 | 323 ± 6 | 0.01 ± <LOD β | 99.82 |
| 100 | 5.6 | 53 ± 1 | 345 ± 26 | 0.11 ± 0.07 | 98.03 |
| 150 | 5.6 | 54 ± 1 | 337 ± 13 | 0.65 ± 0.07 | 88.39 |
| Catalyst Concentration (mmol L−1) | Degradation of IER (%) | TOC Removal (%) |
|---|---|---|
| 25 | 62–75 | 58–78 |
| 50 | 92–100 | 94–99 |
| 100 | 97–99 | 83–98 |
| 150 | 87–93 | 86–94 |
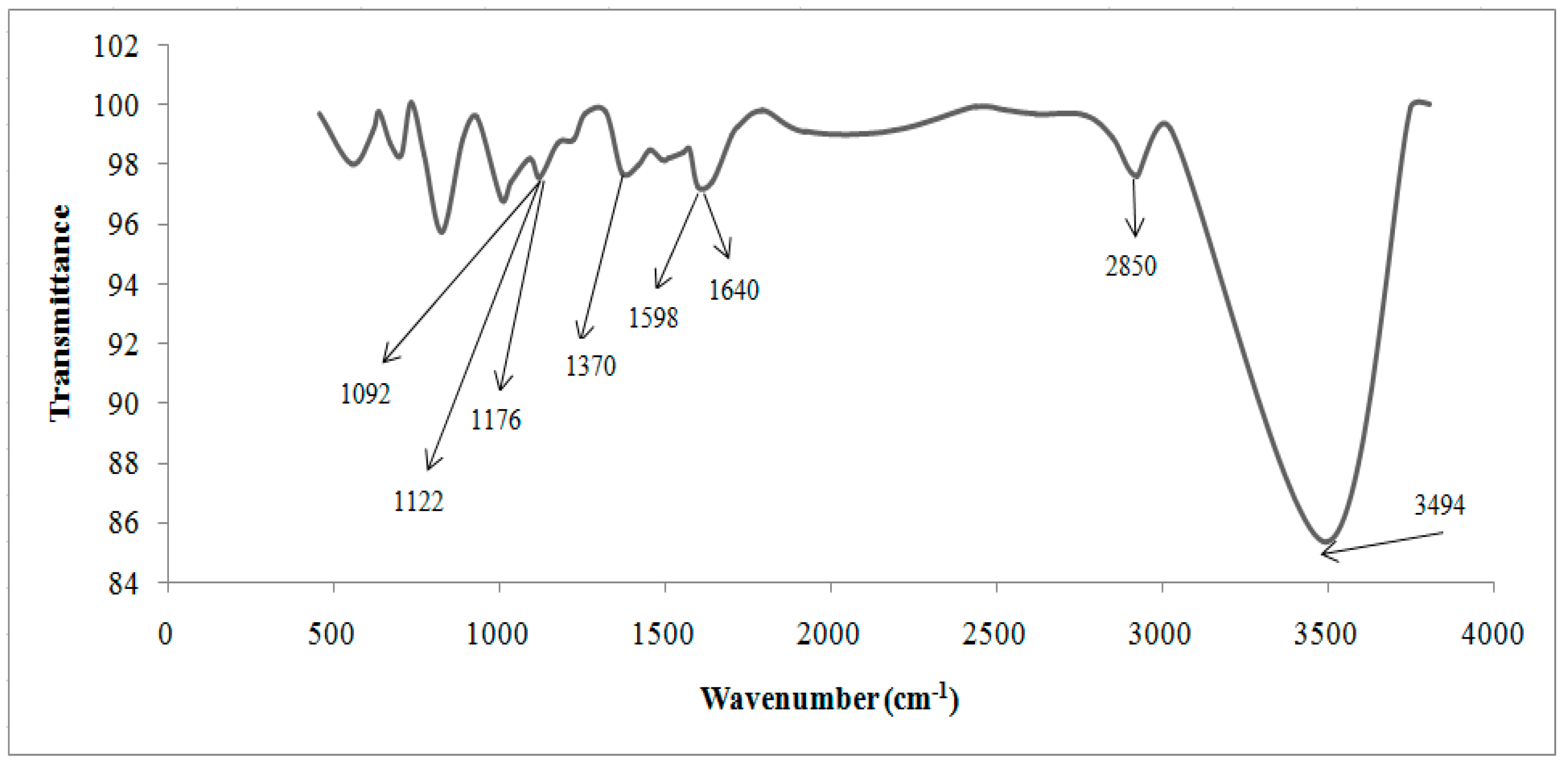
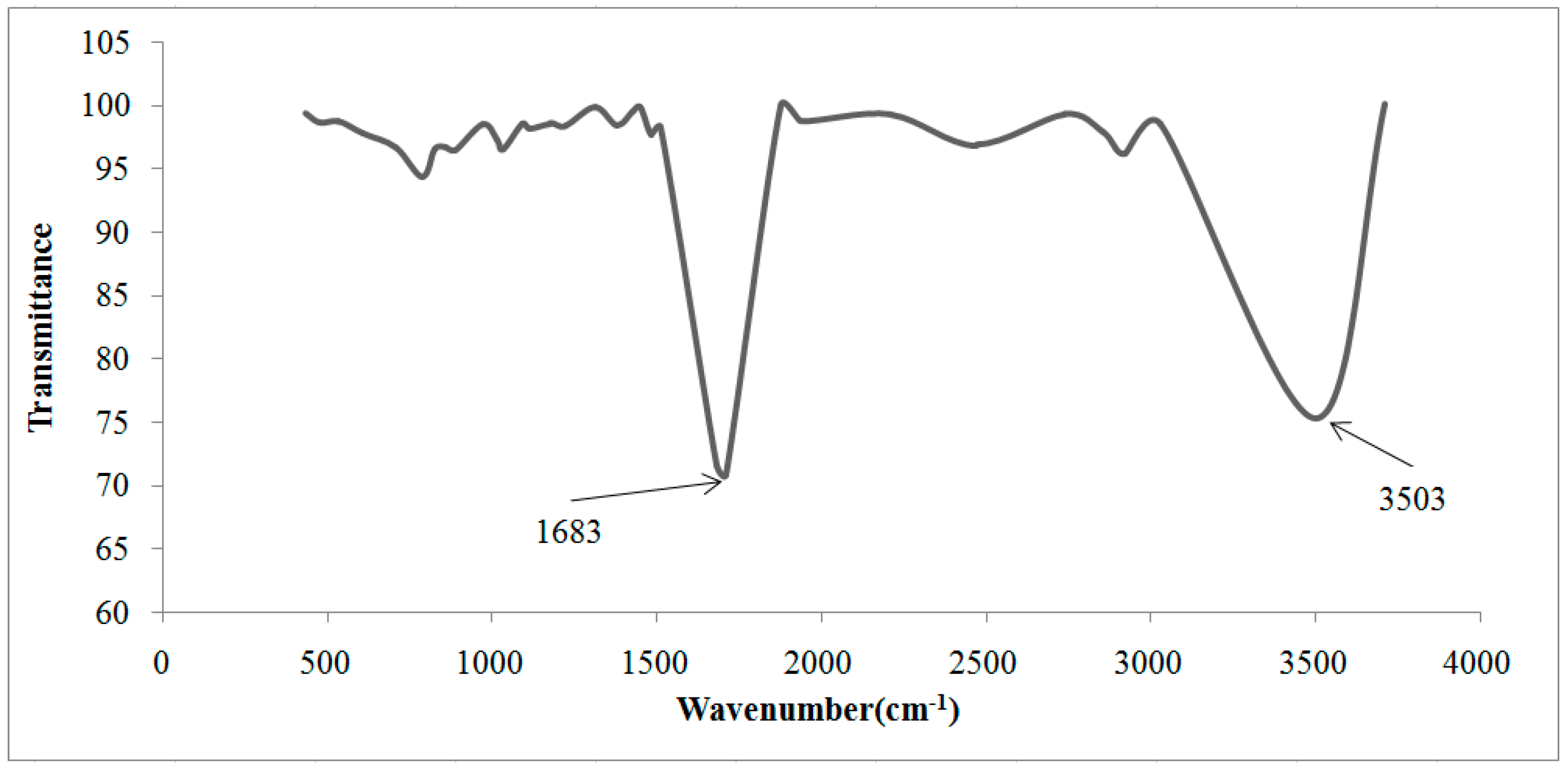
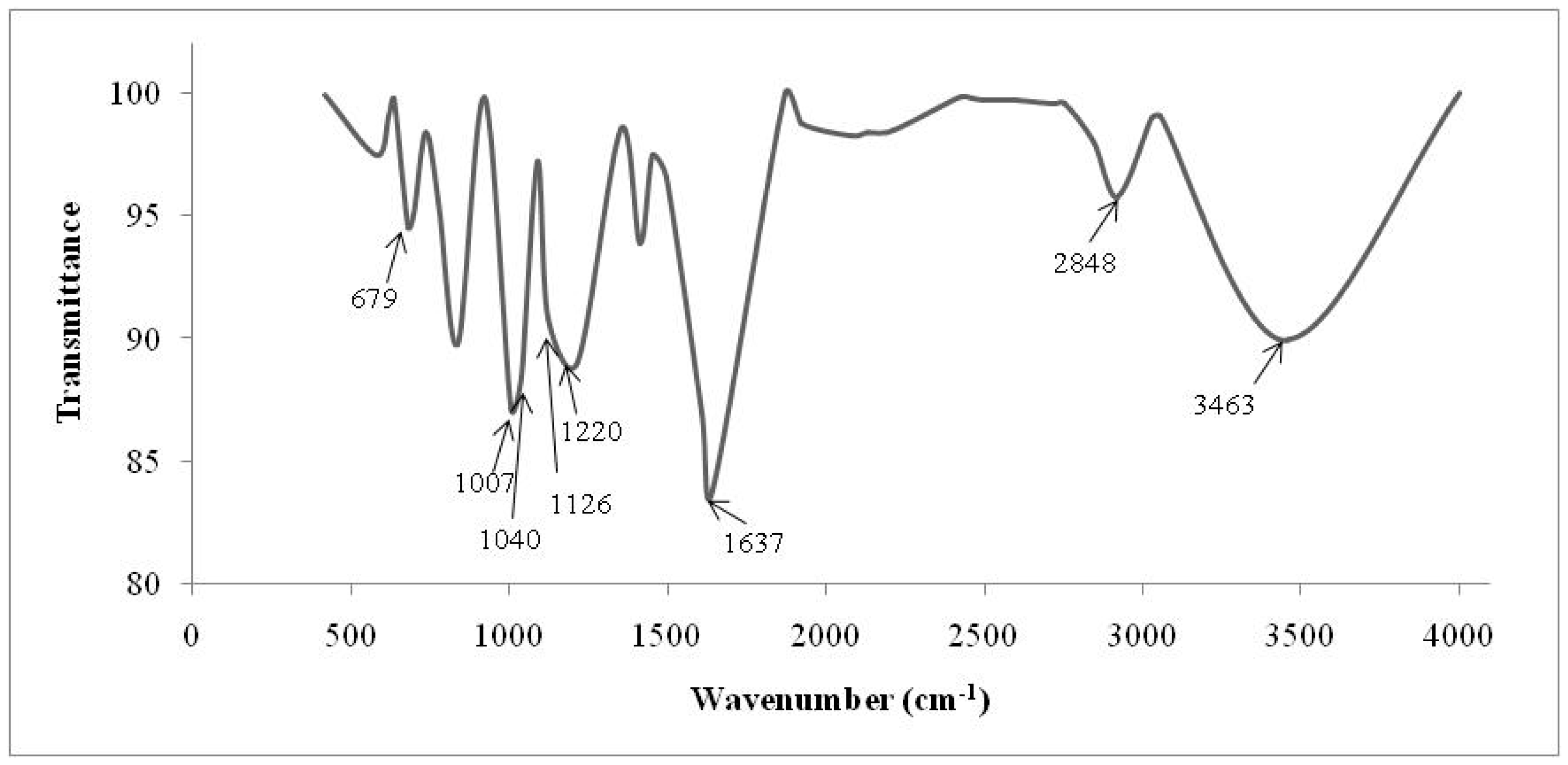
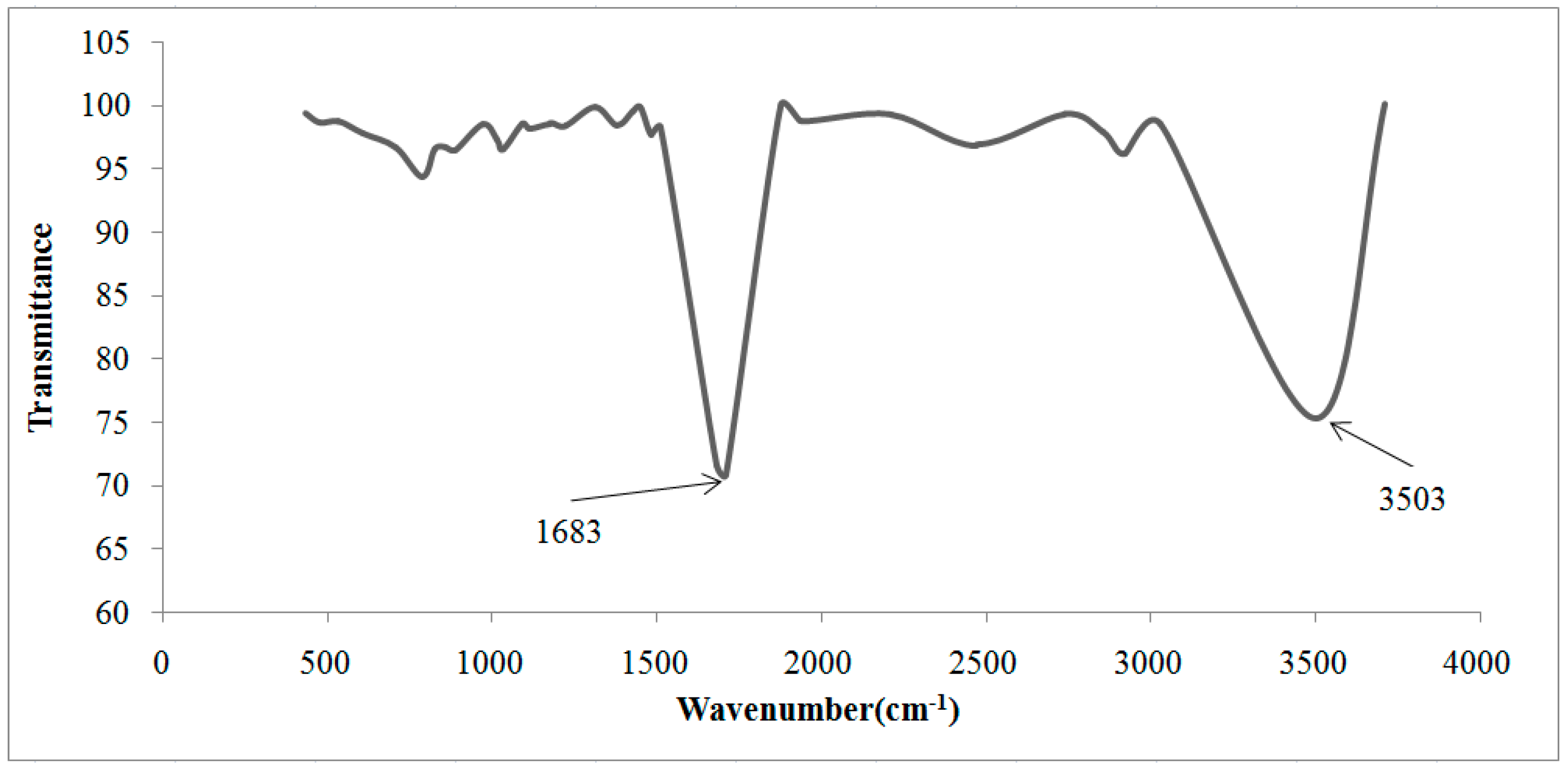
| Cationic without Treatment | Anionic without Treatment | Residual Resins (Cationic and Anionic Resins) | Attribution |
|---|---|---|---|
| 3463 S | 3494 S | 3503 S | O–H water |
| 2848 m | 2850 m | - | Stretch C–H from alkanes |
| - | - | 1683 S | Carboxylate ions |
| 1637 S | 1640 w | - | Stretch of the benzene rings from water hydration |
| - | 1598 w | - | Bond N–H |
| - | 1370 m | - | N–CH3 of quaternaries amine |
| 1220 S, 1126 S, 1040 S and 679 S | - | - | Sulfonic acid |
| - | 1176 m | - | C–N from aliphatic quaternary amines |
| 1126 Sh 1040 S, 1007 S | - | - | SO3 symmetric stretch |
| - | 1122 S 1092 S | - | Aliphatic amines (usually they are shown in double peaks) |
| pH | Setting Time (hours) | Axial Compressive Strength (MPa) * |
|---|---|---|
| Acid | 8 | 16 ± 1 |
| Neutral | 5 | 8 ± 2 |
| Basic | 6 | 8 ± 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Araujo, L.G.; Marumo, J.T. Reaction of Ion Exchange Resins with Fenton’s Reagent. Environments 2018, 5, 123. https://doi.org/10.3390/environments5110123
De Araujo LG, Marumo JT. Reaction of Ion Exchange Resins with Fenton’s Reagent. Environments. 2018; 5(11):123. https://doi.org/10.3390/environments5110123
Chicago/Turabian StyleDe Araujo, Leandro Goulart, and Júlio Takehiro Marumo. 2018. "Reaction of Ion Exchange Resins with Fenton’s Reagent" Environments 5, no. 11: 123. https://doi.org/10.3390/environments5110123
APA StyleDe Araujo, L. G., & Marumo, J. T. (2018). Reaction of Ion Exchange Resins with Fenton’s Reagent. Environments, 5(11), 123. https://doi.org/10.3390/environments5110123