
Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis GI-23 Reveal a Widespread Recombinant Cluster and New Among-Countries Linkages

 , , ,
, , ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. European Dataset
2.2. International Dataset
2.3. Recombination Analysis
2.4. Phylodynamic Analysis
3. Results
3.1. Dataset
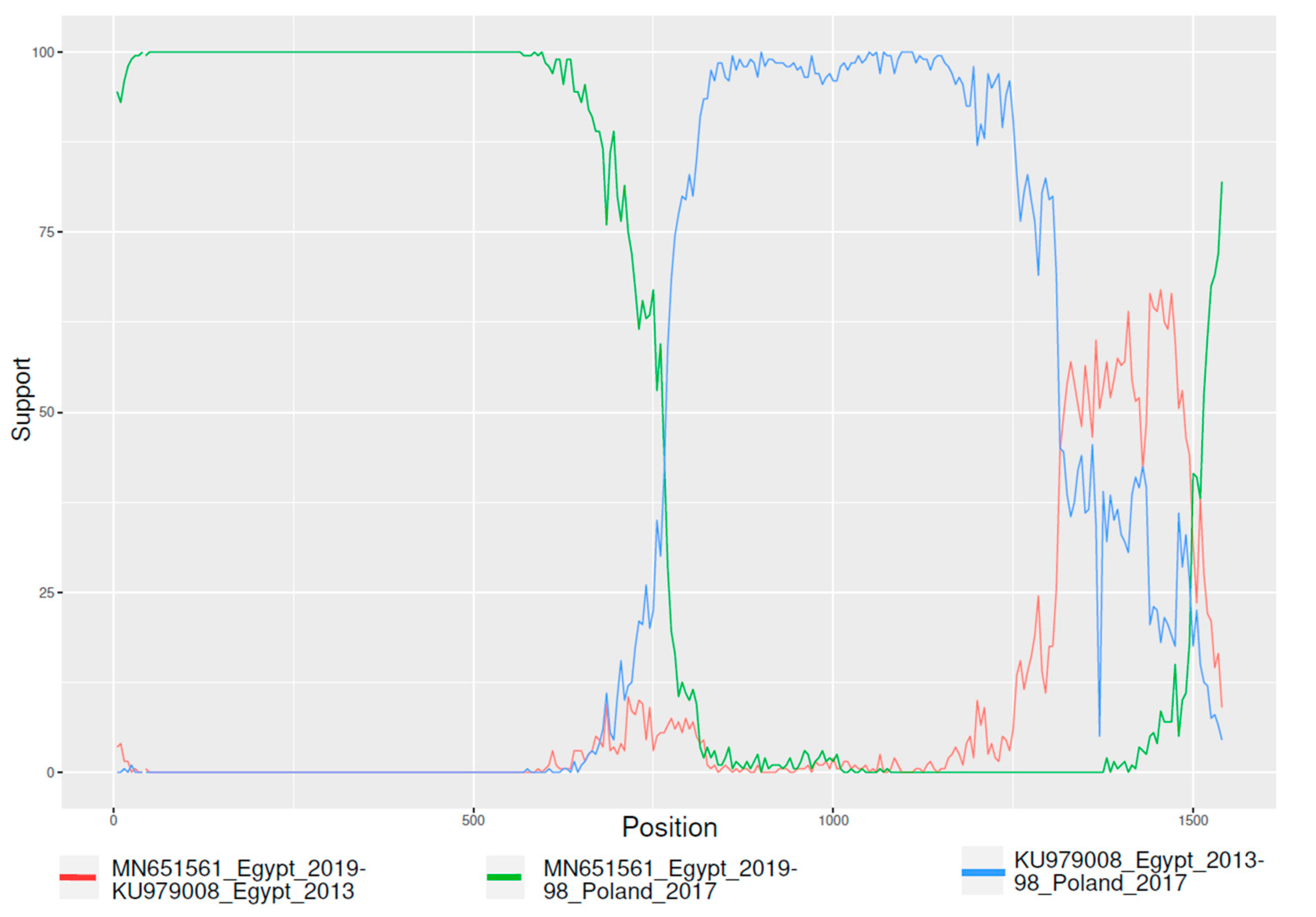
3.2. Recombination Analysis
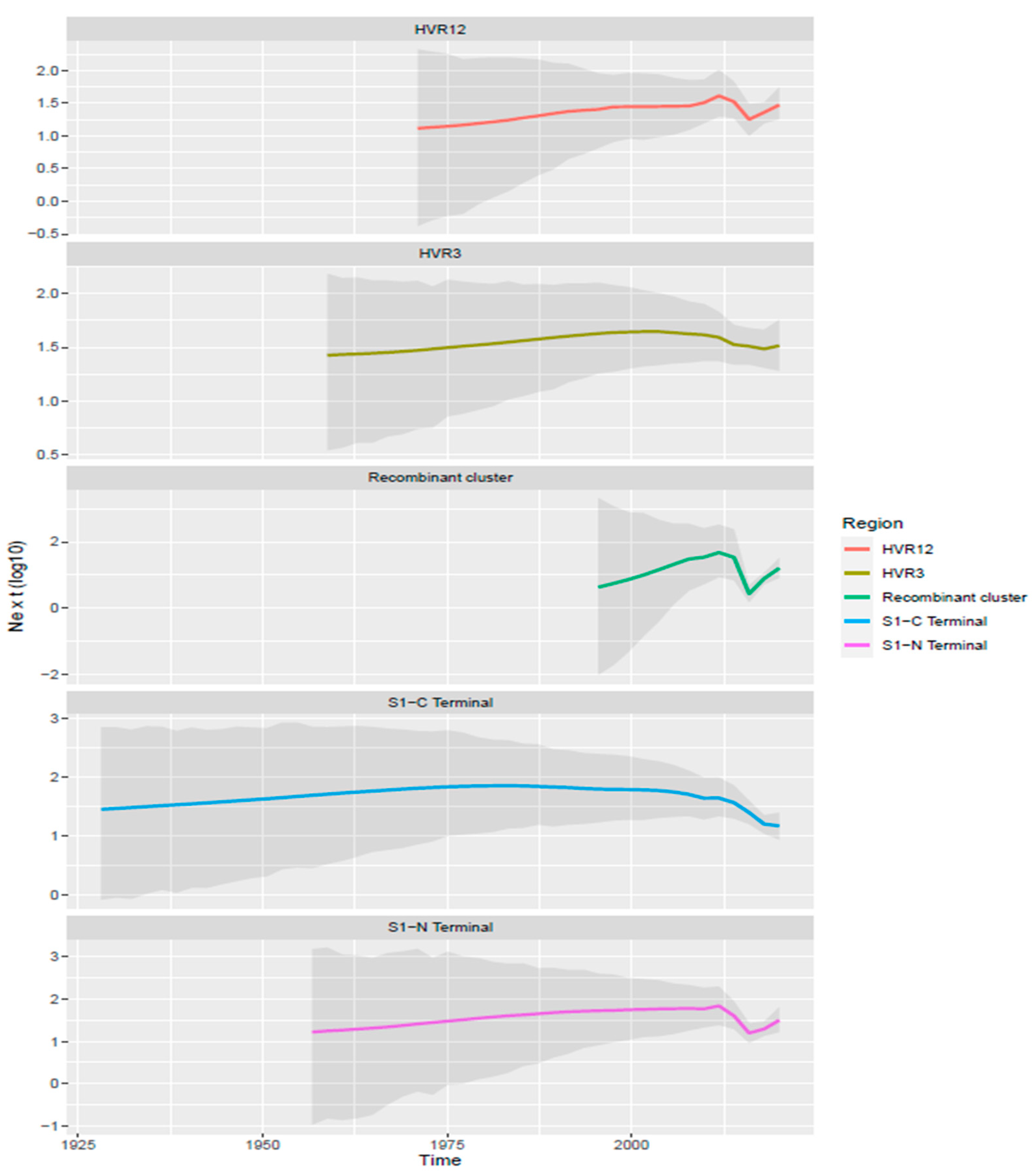
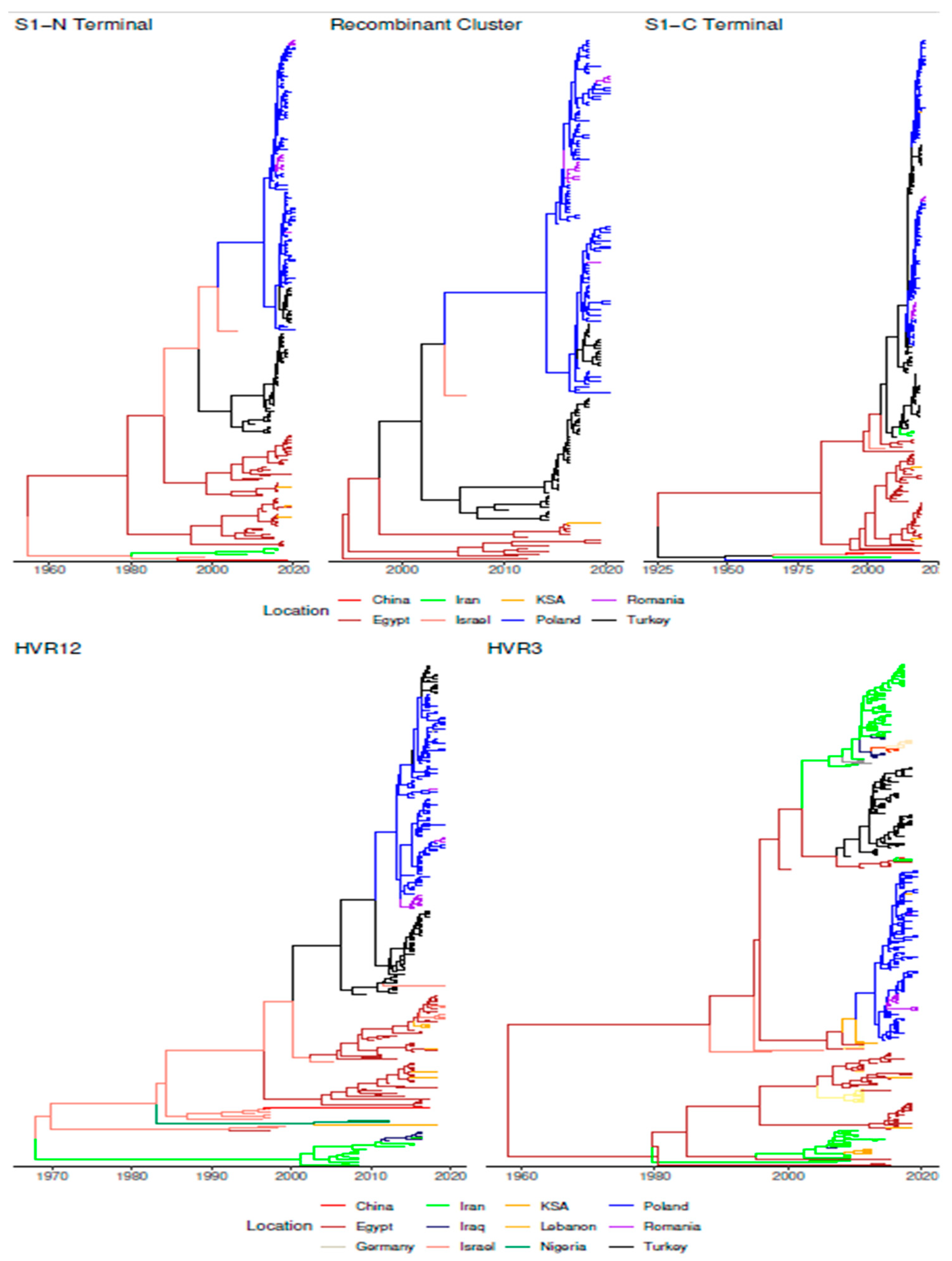
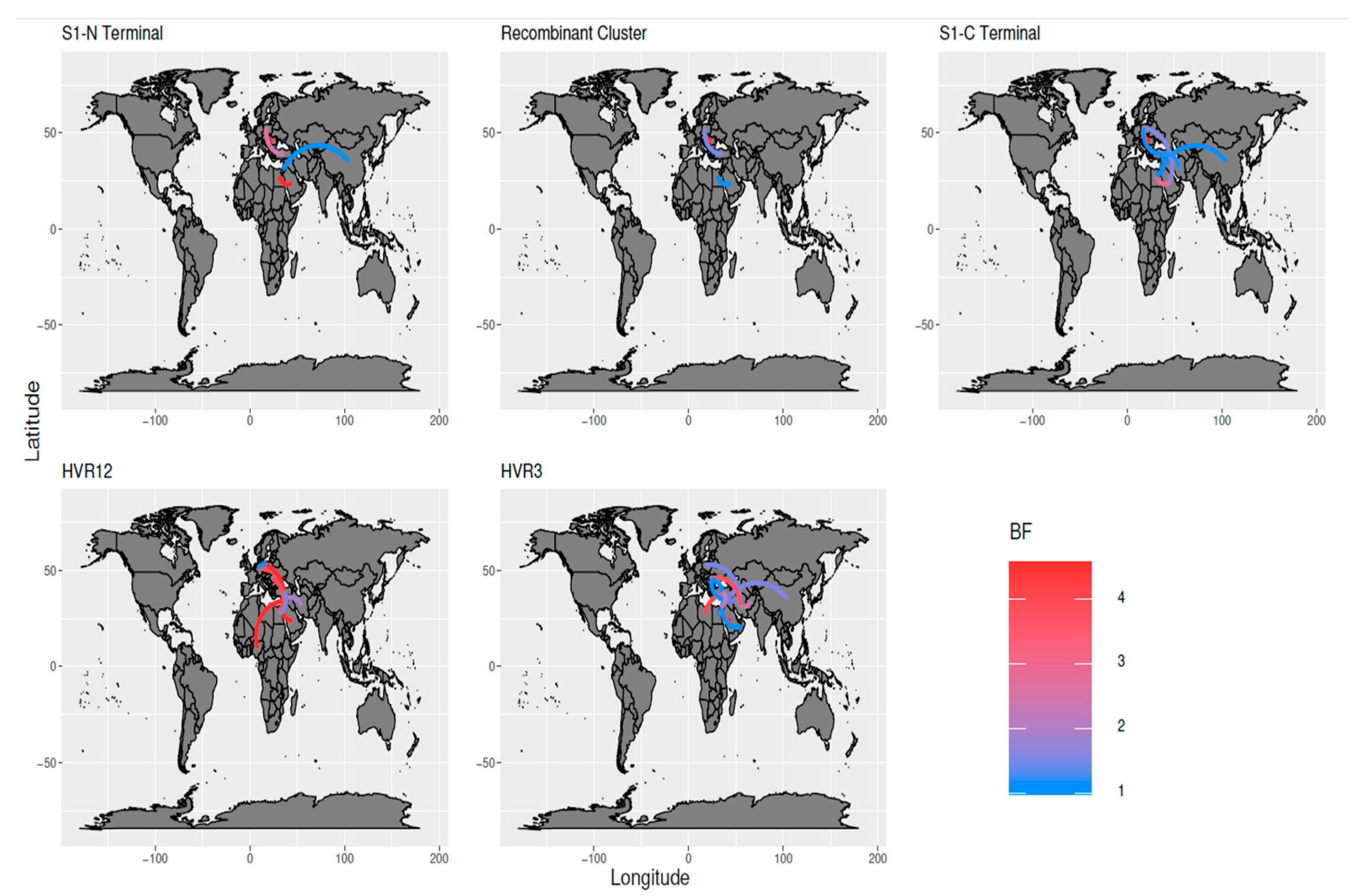
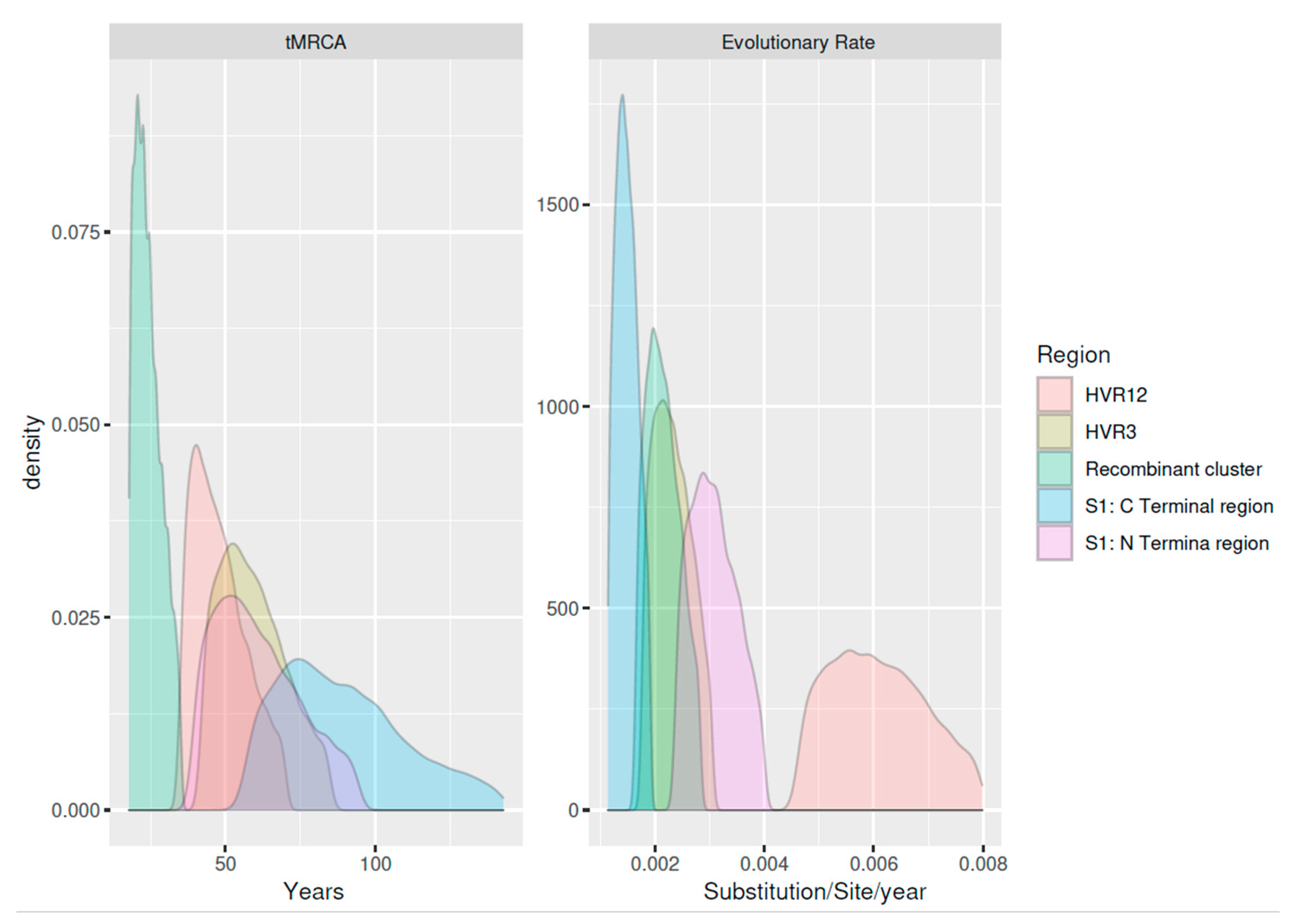
3.3. Phylodynamic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackwood, M.W.; Wit, S. Infectious Bronchitis. In Diseases of Poultry; Wiley: Hoboken, NJ, USA, 2020; Volume 12, pp. 167–188. [Google Scholar]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular Evolution and Emergence of Avian Gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.S. Coronaviridae: Infectious Bronchitis Virus. In Emerging and Re-Emerging Infectious Diseases of Livestock; Bayry, J., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 133–166. ISBN 978-3-319-47426-7. [Google Scholar]
- Cavanagh, D. Coronavirus Avian Infectious Bronchitis Virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global Distributions and Strain Diversity of Avian Infectious Bronchitis Virus: A Review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyeda, Z.; Szeredi, L.; Mató, T.; Süveges, T.; Balka, G.; Abonyi-Tóth, Z.; Rusvai, M.; Palya, V. Comparative Histopathology and Immunohistochemistry of QX-like, Massachusetts and 793/B Serotypes of Infectious Bronchitis Virus Infection in Chickens. J. Comp. Pathol. 2010, 143, 276–283. [Google Scholar] [CrossRef]
- Owen, R.L.; Cowen, B.S.; Hattel, A.L.; Naqi, S.A.; Wilson, R.A. Detection of Viral Antigen Following Exposure of One-Day-Old Chickens to the Holland 52 Strain of Infectious Bronchitis Virus. Avian Pathol. 1991, 20, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think Globally, Act Locally: Phylodynamic Reconstruction of Infectious Bronchitis Virus (IBV) QX Genotype (GI-19 Lineage) Reveals Different Population Dynamics and Spreading Patterns When Evaluated on Different Epidemiological Scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef]
- Moreno, A.; Franzo, G.; Massi, P.; Tosi, G.; Blanco, A.; Antilles, N.; Biarnes, M.; Majó, N.; Nofrarías, M.; Dolz, R.; et al. A Novel Variant of the Infectious Bronchitis Virus Resulting from Recombination Events in Italy and Spain. Avian Pathol. 2017, 46, 28–35. [Google Scholar] [CrossRef]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 Gene-Based Phylogeny of Infectious Bronchitis Virus: An Attempt to Harmonize Virus Classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef]
- Cavanagh, D.; Ellis, M.M.; Cook, J.K.A. Relationship between Sequence Variation in the S1 Spike Protein of Infectious Bronchitis Virus and the Extent of Cross-Protection in Vivo. Avian Pathol. 1997, 26, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zhao, W.; Han, Z.; Chen, Y.; Zhao, Y.; Sun, J.; Li, H.; Shao, Y.; Liu, L.; Liu, S. Genome Characterization, Antigenicity and Pathogenicity of a Novel Infectious Bronchitis Virus Type Isolated from South China. Infect. Genet. Evol. 2017, 54, 437–446. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, L.; Zhao, W.; Liu, L.; Zhao, Y.; Shao, Y.; Li, H.; Han, Z.; Liu, S. Identification and Molecular Characterization of a Novel Serotype Infectious Bronchitis Virus (GI-28) in China. Vet. Microbiol. 2017, 198, 108–115. [Google Scholar] [CrossRef]
- Ma, T.; Xu, L.; Ren, M.; Shen, J.; Han, Z.; Sun, J.; Zhao, Y.; Liu, S. Novel Genotype of Infectious Bronchitis Virus Isolated in China. Vet. Microbiol. 2019, 230, 178–186. [Google Scholar] [CrossRef]
- Domanska-Blicharz, K.; Sajewicz-Krukowska, J.; Lisowska, A. New PA/1220/98-like Variant of Infectious Bronchitis Virus in Poland. Avian Pathol. 2020, 49, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moneim, A.S.; Madbouly, H.M.; Gelb, J.; Ladman, B.S. Isolation and Identification of Egypt/Beni-Seuf/01 a Novel Genotype of Infectious Bronchitis Virus. Vet. Med J. Giza 2002, 50, 1065–1078. [Google Scholar]
- Meir, R.; Rosenblut, E.; Perl, S.; Kass, N.; Ayali, G.; Hemsani, E.; Perk, S. Identification of a Novel Nephropathogenic Infectious Bronchitis Virus in Israel. Avian Dis. 2004, 48, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Houta, M.H.; Hassan, K.E.; El-Sawah, A.A.; Elkady, M.F.; Kilany, W.H.; Ali, A.; Abdel-Moneim, A.S. The Emergence, Evolution and Spread of Infectious Bronchitis Virus Genotype GI-23. Arch. Virol. 2021, 166, 9–26. [Google Scholar] [CrossRef]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral Phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [Green Version]
- Standley, K. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.(Outlines Version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Biol. Evol. 2013, 30, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Klosterhalfen, D.; Kump, F.W.S.; Casteel, M. Research Note: First Evidence of Infectious Bronchitis Virus Middle-East GI-23 Lineage (Var2-like) in Germany. Poult. Sci. 2020, 99, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, A.; Sajewicz-Krukowska, J.; Fusaro, A.; Pikula, A.; Domanska-Blicharz, K. First Characterization of a Middle-East GI-23 Lineage (Var2-like) of Infectious Bronchitis Virus in Europe. Virus Res. 2017, 242, 43–48. [Google Scholar] [CrossRef]
- Mahmood, Z.H.; Sleman, R.R.; Uthman, A.U. Isolation and Molecular Characterization of Sul/01/09 Avian Infectious Bronchitis Virus, Indicates the Emergence of a New Genotype in the Middle East. Vet. Microbiol. 2011, 150, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sadri, N.; Ghalyanchilangeroudi, A.; Mehrabadi, M.H.F.; Hosseini, H.; Shayeganmehr, A.; Sediqian, M.S.; Jabbarifakhr, M.; Hamdan, A.M.; Mousavi, F.S. Genotyping of Avian Infectious Bronchitis Virus in Afghanistan (2016–2017): The First Report. Iran. J. Vet. Res. 2019, 20, 60–63. [Google Scholar]
- Franzo, G.; Cecchinato, M.; Tosi, G.; Fiorentini, L.; Faccin, F.; Tucciarone, C.M.; Trogu, T.; Barbieri, I.; Massi, P.; Moreno, A. GI-16 lineage (624/I or Q1), there and back again: The history of one of the major threats for poultry farming of our era. PLoS ONE 2018, 13, e0203513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of Evolutionary Change in Viruses: Patterns and Determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Al-Mubarak, A.I.A.; Al-Kubati, A.A.G. Cocirculation of Four Infectious Bronchitis Virus Lineages in Broiler Chickens in the Eastern Region of Saudi Arabia from 2012 to 2014. Vet. Med. Int. 2020, 2020, 6037893. [Google Scholar] [CrossRef] [PubMed]
- Gelb, J.; Weisman, Y.; Ladman, B.S.; Meir, R. S1 Gene Characteristics and Efficacy of Vaccination against Infectious Bronchitis Virus Field Isolates from the United States and Israel (1996 to 2000). Avian Pathol. 2005, 34, 194–203. [Google Scholar] [CrossRef]
- Hosseini, H.; Fard, M.H.B.; Charkhkar, S.; Morshed, R. Epidemiology of Avian Infectious Bronchitis Virus Genotypes in Iran (2010–2014). Avian Dis. 2015, 59, 431–435. [Google Scholar] [CrossRef]
- Moharam, I.; Sultan, H.; Hassan, K.; Ibrahim, M.; Shany, S.; Shehata, A.A.; Abo-ElKhair, M.; Pfaff, F.; Höper, D.; el Kady, M.; et al. Emerging Infectious Bronchitis Virus (IBV) in Egypt: Evidence for an Evolutionary Advantage of a New S1 Variant with a Unique Gene 3ab Constellation. Infect. Genet. Evol. 2020, 85, 104433. [Google Scholar] [CrossRef] [PubMed]
- Zanaty, A.; Naguib, M.M.; El-Husseiny, M.H.; Mady, W.; Hagag, N.; Arafa, A.S. The Sequence of the Full Spike S1 Glycoprotein of Infectious Bronchitis Virus Circulating in Egypt Reveals Evidence of Intra-Genotypic Recombination. Arch. Virol. 2016, 161, 3583–3587. [Google Scholar] [CrossRef] [PubMed]
- Kahya, S.; Coven, F.; Temelli, S.; Eyigor, A.; Carli, K.T. Presence of IS/1494/06 Genotype-Related Infectious Bronchitis Virus in Breeder and Broiler Flocks in Turkey. Ank. Univ. Vet. Fak. Derg. 2012, 60, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, H.; Altan, E.; Cizmecigil, U.Y.; Gurel, A.; Ozturk, G.Y.; Bamac, O.E.; Aydin, O.; Britton, P.; Monne, I.; Cetinkaya, B.; et al. Phylogeny and S1 Gene Variation of Infectious Bronchitis Virus Detected in Broilers and Layers in Turkey. Avian Dis. 2016, 60, 596–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelwhab, E.M.; Hassan, M.K.; Abdel-Moneim, A.S.; Naguib, M.M.; Mostafa, A.; Hussein, I.T.M.; Arafa, A.; Erfan, A.M.; Kilany, W.H.; Agour, M.G.; et al. Introduction and Enzootic of A/H5N1 in Egypt: Virus Evolution, Pathogenicity and Vaccine Efficacy Ten Years On. Infect. Genet. Evol. 2016, 40, 80–90. [Google Scholar] [CrossRef]
- Abozeid, H.H.; Paldurai, A.; Varghese, B.P.; Khattar, S.K.; Afifi, M.A.; Zouelfakkar, S.; El-Deeb, A.H.; El-Kady, M.F.; Samal, S.K. Development of a Recombinant Newcastle Disease Virus-Vectored Vaccine for Infectious Bronchitis Virus Variant Strains Circulating in Egypt. Vet. Res. 2019, 50, 12. [Google Scholar] [CrossRef]
- Al-Ebshahy, E.; Abdel-Sabour, M.; Abas, O.; Yanai, T. Protection Conferred by a Vaccine Derived from an Inactivated Egyptian Variant of Infectious Bronchitis Virus: A Challenge Experiment. Trop. Anim. Health Prod. 2019, 51, 1997–2001. [Google Scholar] [CrossRef]
- Ali, A.; Kilany, W.H.; El-Abideen, M.A.Z.; el Sayed, M.; Elkady, M. Safety and Efficacy of Attenuated Classic and Variant 2 Infectious Bronchitis Virus Candidate Vaccines. Poult. Sci. 2018, 97, 4238–4244. [Google Scholar] [CrossRef]
- Sultan, H.A.; Ali, A.; el Feil, W.K.; Bazid, A.H.I.; Zain El-Abideen, M.A.; Kilany, W.H. Protective Efficacy of Different Live Attenuated Infectious Bronchitis Virus Vaccination Regimes Against Challenge with IBV Variant-2 Circulating in the Middle East. Front. Vet. Sci. 2019, 6, 341. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Tucciarone, C.M.; Drigo, M.; Cecchinato, M.; Enache, M.; Bejan, V. Infectious Bronchitis Virus in Romanian Broiler Farms. Vet. Rec. 2017, 180, 574. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.S.; Afifi, M.A.; El-Kady, M.F. Emergence of a Novel Genotype of Avian Infectious Bronchitis Virus in Egypt. Arch. Virol. 2012, 157, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, A.; Pikuła, A.; Opolska, J.; Jasik, A.; Kycko, A.; Domańska-Blicharz, K. Virulence Properties of Gi-23 Infectious Bronchitis Virus Isolated in Poland and Efficacy of Different Vaccination Strategies. Pathogens 2021, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Amin, O.G.M.; Valastro, V.; Salviato, A.; Drago, A.; Cattoli, G.; Monne, I. Circulation of QX-like Infectious Bronchitis Virus in the Middle East. Vet. Rec. 2012, 171, 530. [Google Scholar] [CrossRef]
- Seger, W.; GhalyanchiLangeroudi, A.; Karimi, V.; Madadgar, O.; Marandi, M.V.; Hashemzadeh, M. Genotyping of Infectious Bronchitis Viruses from Broiler Farms in Iraq during 2014-2015. Arch. Virol. 2016, 161, 1229–1237. [Google Scholar] [CrossRef]
- Rohaim, M.A.; el Naggar, R.F.; Helal, A.M.; Bayoumi, M.M.; El-Saied, M.A.; Ahmed, K.A.; Shabbir, M.Z.; Munir, M. Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds. Viruses 2019, 11, 57. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
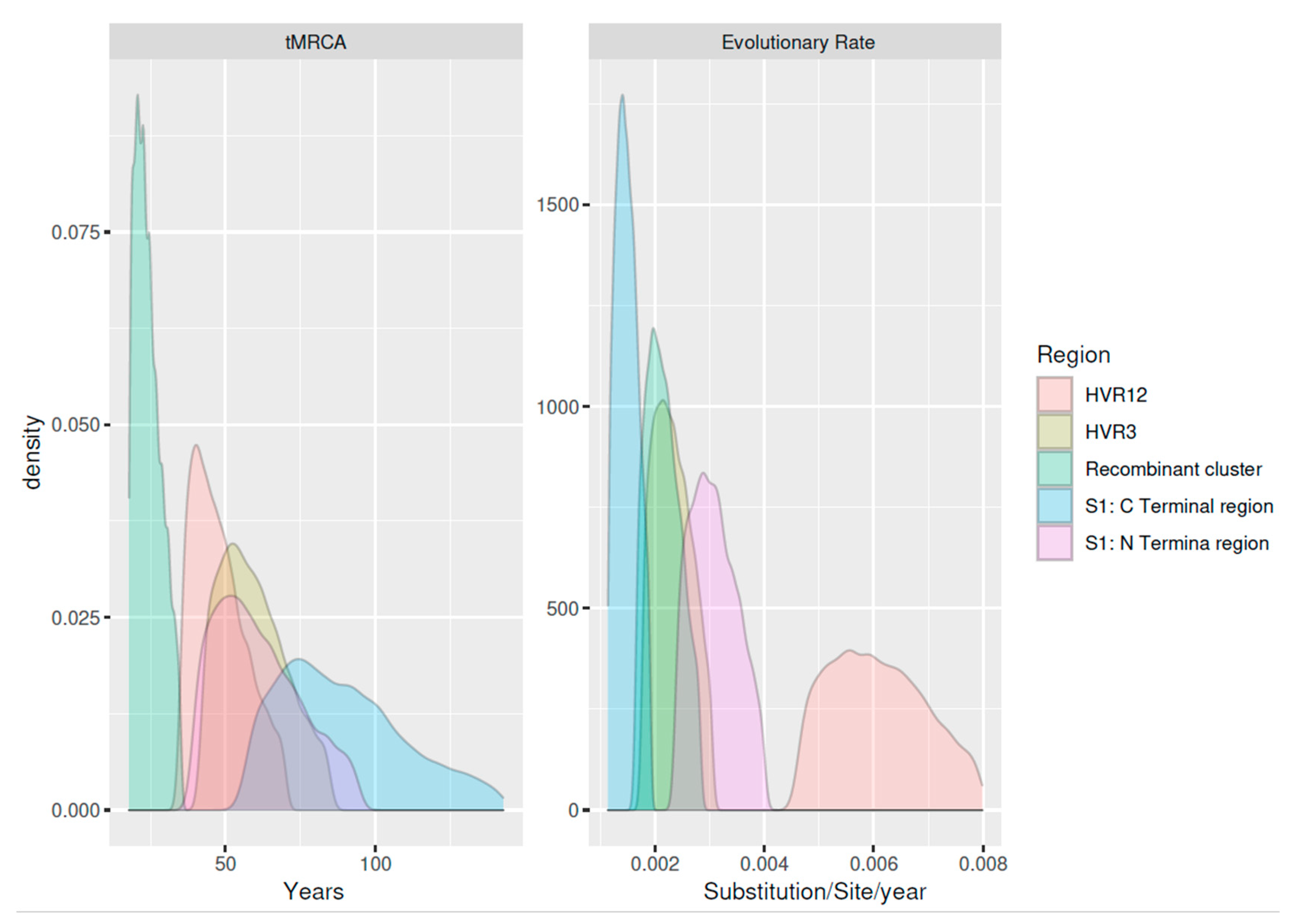
| Time to the Most Recent Common Ancestor (Years) | Origin | Evolutionary Rate | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Database | Included Sequences | Sequence Number | Years | Country Number | Mean | Lower 95HPD | Higher 95HPD | Mean | Lower 95HPD | Higher 95HPD | Mean | Lower 95HPD | Higher 95HPD |
| A | S1-N terminal | 231 | 1998–2020 | 8 | 58.737 | 43.828 | 81.972 | 1961.263 | 1938.028 | 1976.172 | 3.05 × 10−3 | 2.54 × 10−3 | 3.69 × 10−3 |
| B | S1-C terminal | 231 | 1998–2020 | 8 | 86.130 | 64.250 | 120.229 | 1933.87 | 1899.771 | 1955.75 | 1.48 × 10−3 | 1.23 × 10−3 | 1.78 × 10−3 |
| C | Recombinant cluster | 188 | 2006–2020 | 6 | 23.618 | 19.008 | 31.081 | 1996.382 | 1988.919 | 2000.992 | 1.77 × 10−3 | 2.13 × 10−3 | 2.60 × 10−3 |
| D | HVR1/2 | 270 | 1996–2020 | 12 | 57.942 | 45.787 | 75.2 | 1962.058 | 1944.8 | 1974.213 | 6.02 × 10−3 | 4.96 × 10−3 | 7.33 × 10−3 |
| E | HVR3 | 341 | 1998–2020 | 14 | 46.622 | 37.628 | 61.372 | 1973.378 | 1958.628 | 1982.372 | 1.90 × 10−3 | 2.30 × 10−3 | 2.81 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houta, M.H.; Hassan, K.E.; Legnardi, M.; Tucciarone, C.M.; Abdel-Moneim, A.S.; Cecchinato, M.; El-Sawah, A.A.; Ali, A.; Franzo, G. Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis GI-23 Reveal a Widespread Recombinant Cluster and New Among-Countries Linkages. Animals 2021, 11, 3182. https://doi.org/10.3390/ani11113182
Houta MH, Hassan KE, Legnardi M, Tucciarone CM, Abdel-Moneim AS, Cecchinato M, El-Sawah AA, Ali A, Franzo G. Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis GI-23 Reveal a Widespread Recombinant Cluster and New Among-Countries Linkages. Animals. 2021; 11(11):3182. https://doi.org/10.3390/ani11113182
Chicago/Turabian StyleHouta, Mohamed H., Kareem E. Hassan, Matteo Legnardi, Claudia M. Tucciarone, Ahmed S. Abdel-Moneim, Mattia Cecchinato, Azza A. El-Sawah, Ahmed Ali, and Giovanni Franzo. 2021. "Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis GI-23 Reveal a Widespread Recombinant Cluster and New Among-Countries Linkages" Animals 11, no. 11: 3182. https://doi.org/10.3390/ani11113182
APA StyleHouta, M. H., Hassan, K. E., Legnardi, M., Tucciarone, C. M., Abdel-Moneim, A. S., Cecchinato, M., El-Sawah, A. A., Ali, A., & Franzo, G. (2021). Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis GI-23 Reveal a Widespread Recombinant Cluster and New Among-Countries Linkages. Animals, 11(11), 3182. https://doi.org/10.3390/ani11113182