
Single Strain High-Depth NGS Reveals High rDNA (ITS-LSU) Variability in the Four Prevalent Pathogenic Species of the Genus Candida

 ,
,  , ,
, , 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
2.2. MALDI-TOF Analysis
2.3. DNA Extraction and Sequencing
2.4. Data Analysis
2.4.1. MALDI-TOF Data Analysis
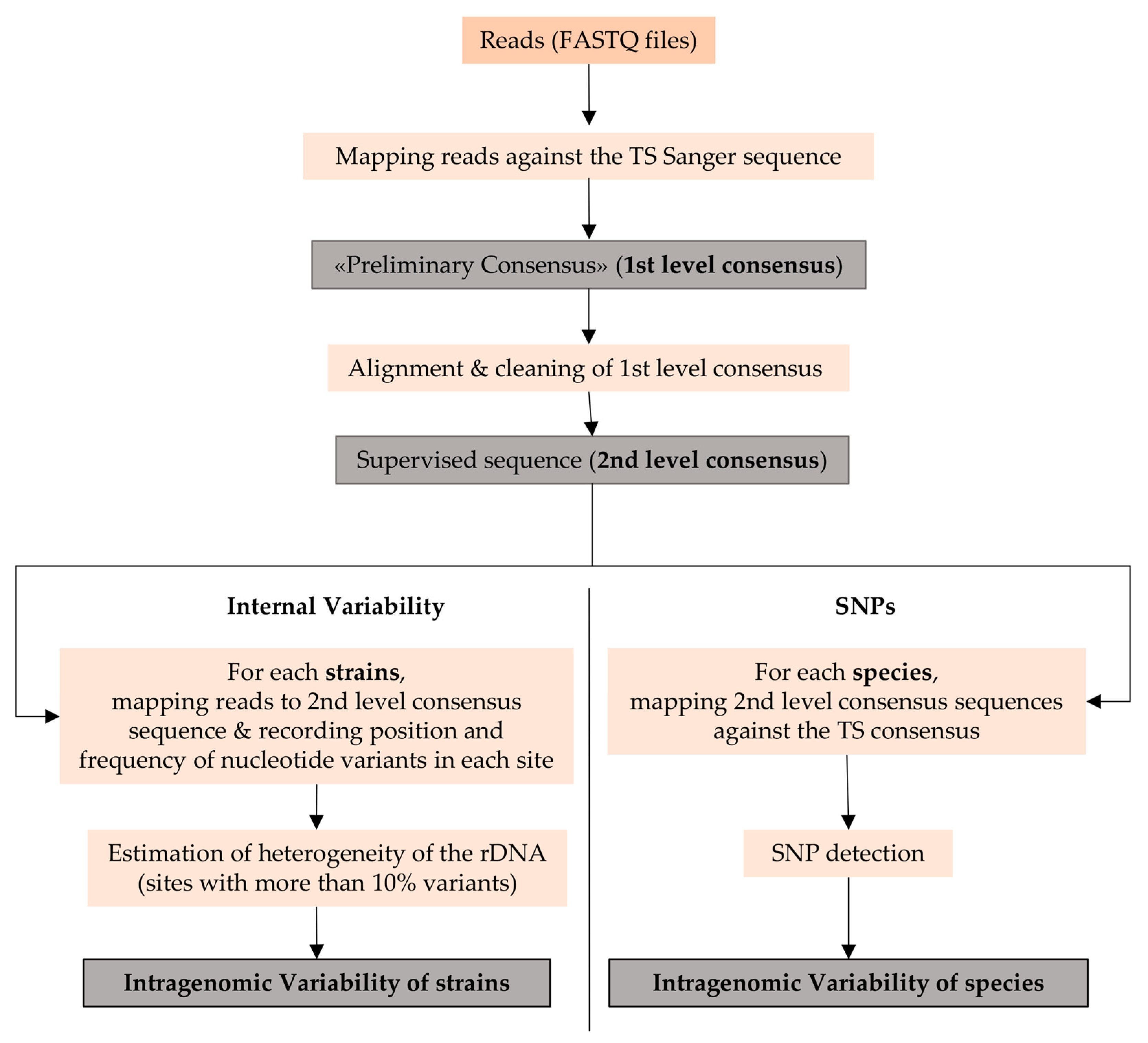
2.4.2. Sequencing Data Analysis
2.4.3. Analysis of Variant Sites
2.4.4. SNP-Based Phylogenetic Analysis
3. Results
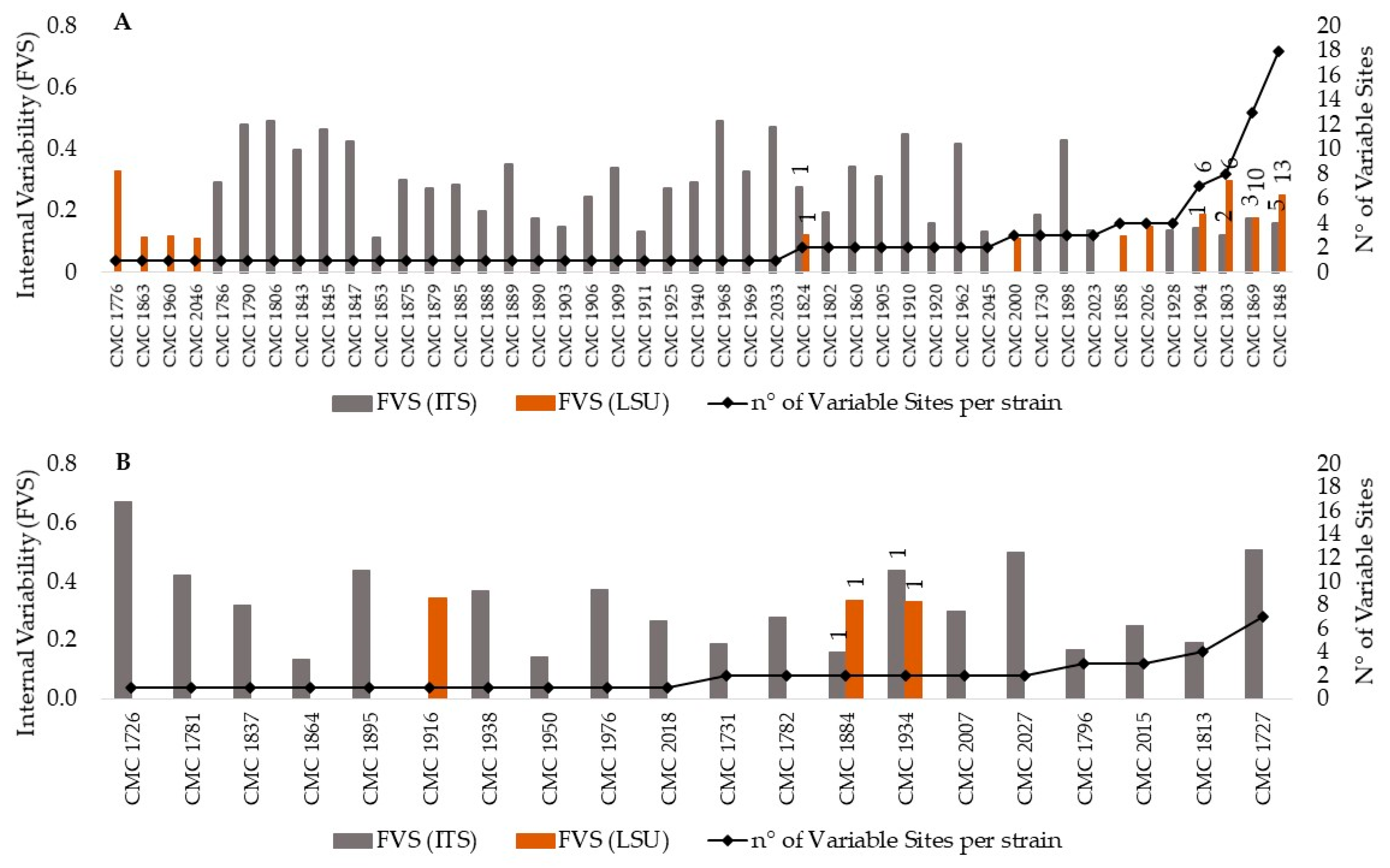
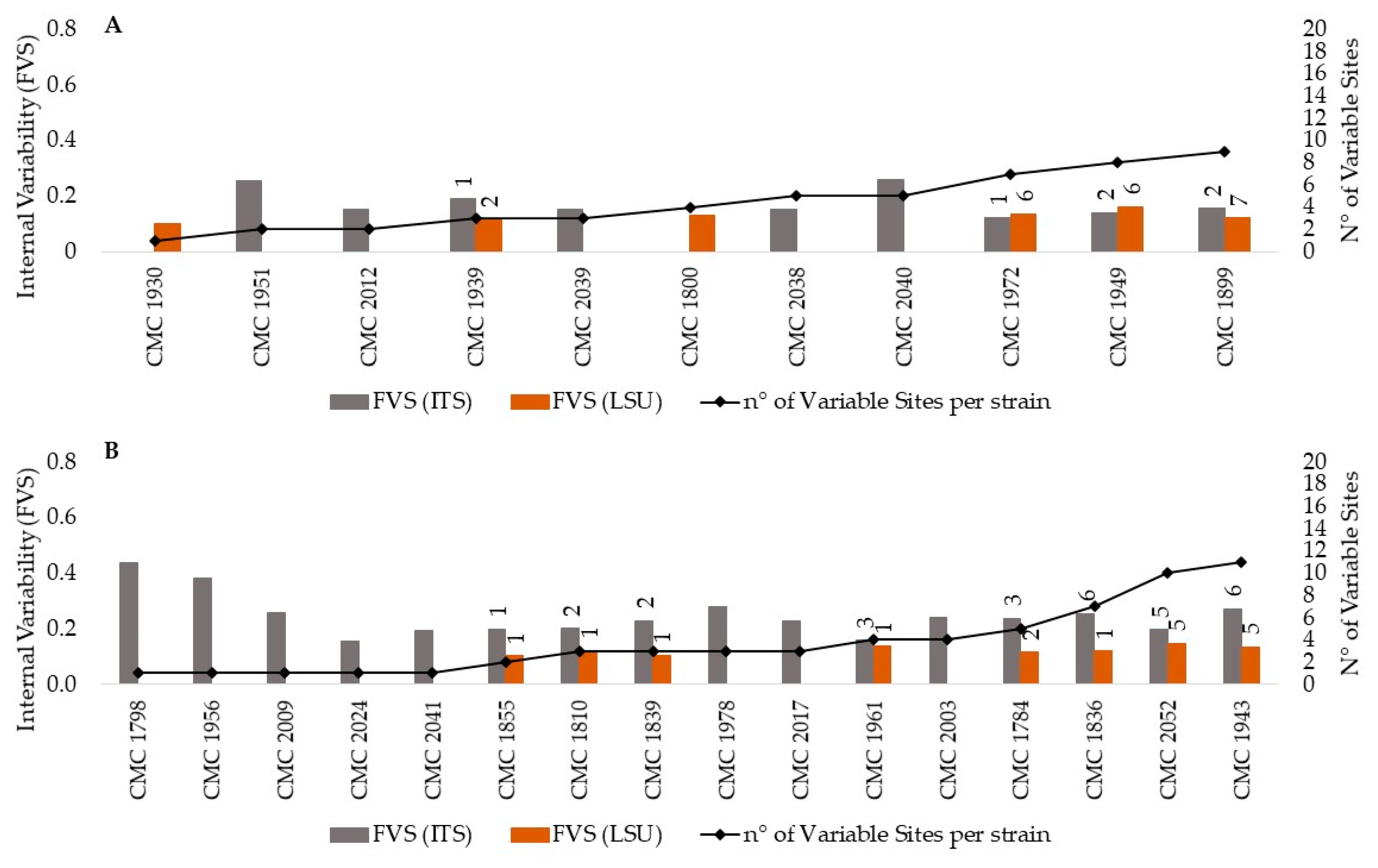
3.1. Heterogeneity of the rDNA within Strains of Candida Species
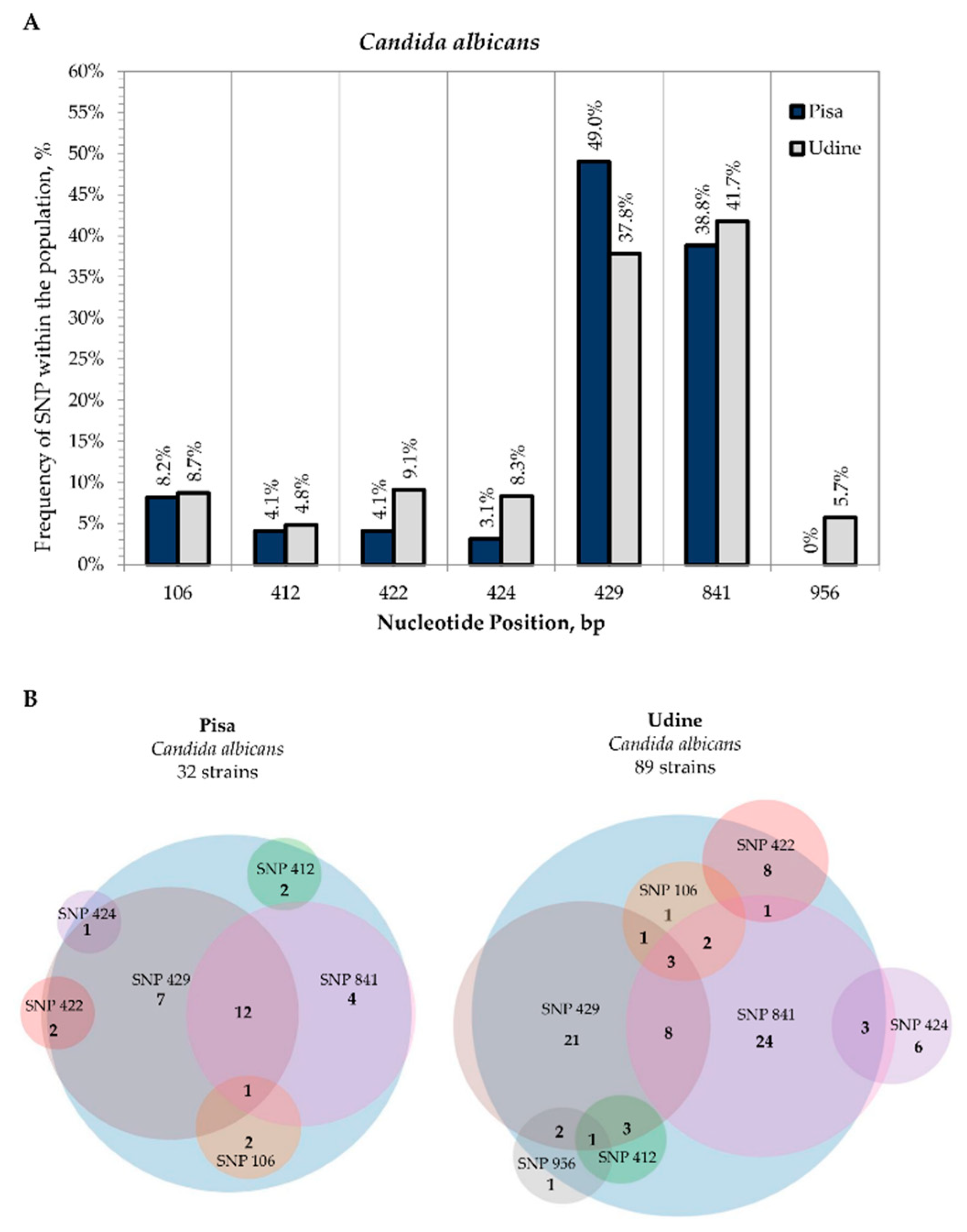
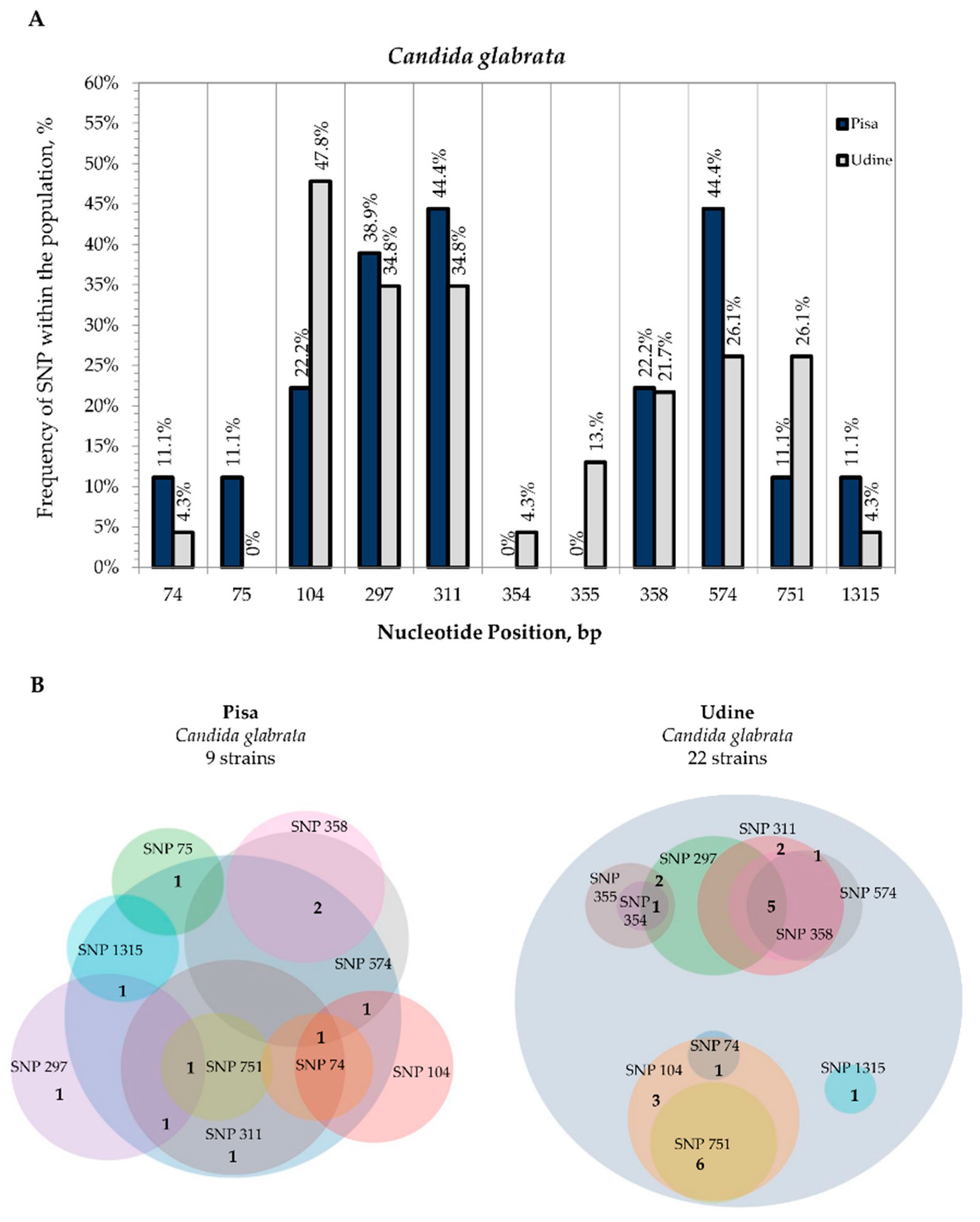
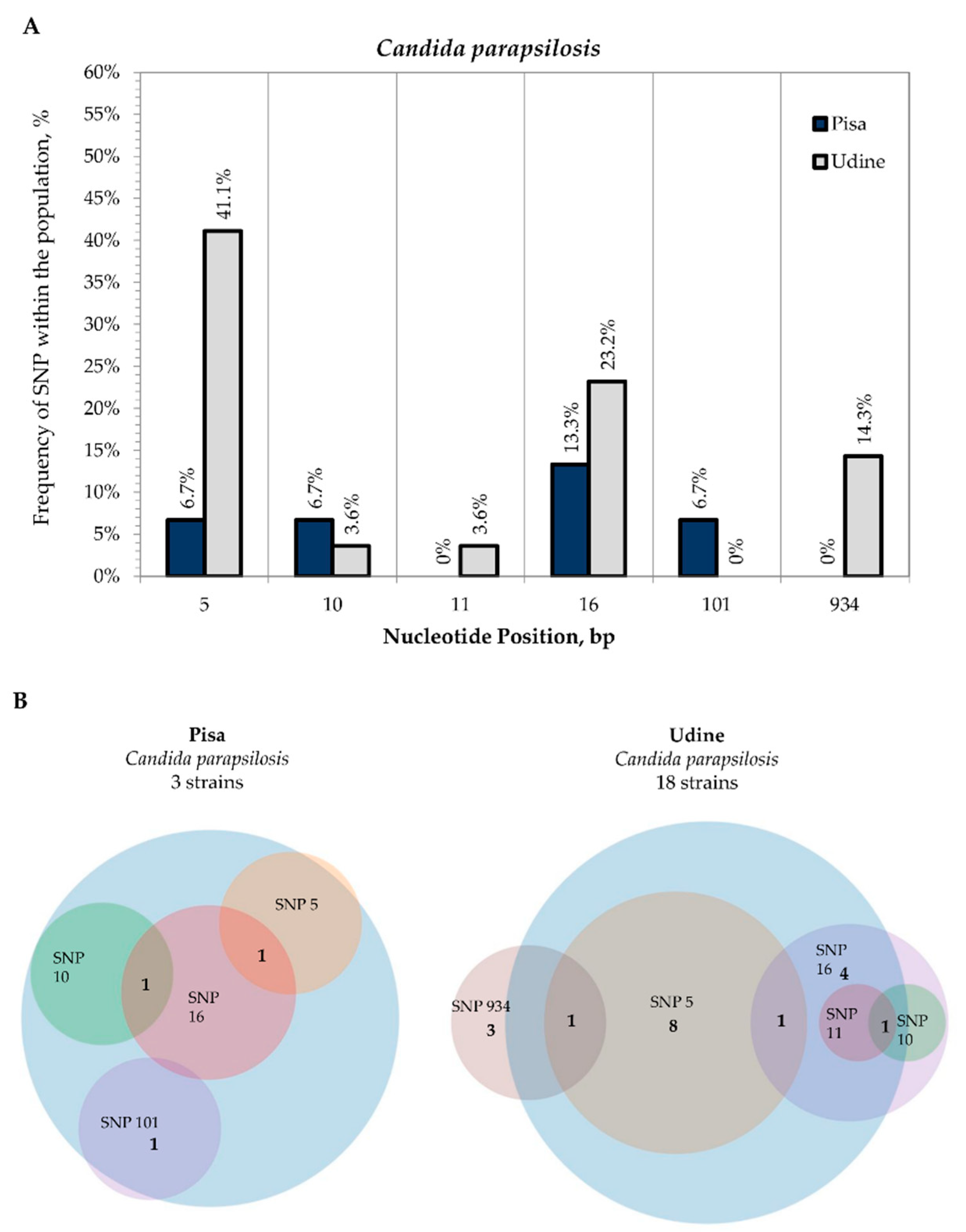
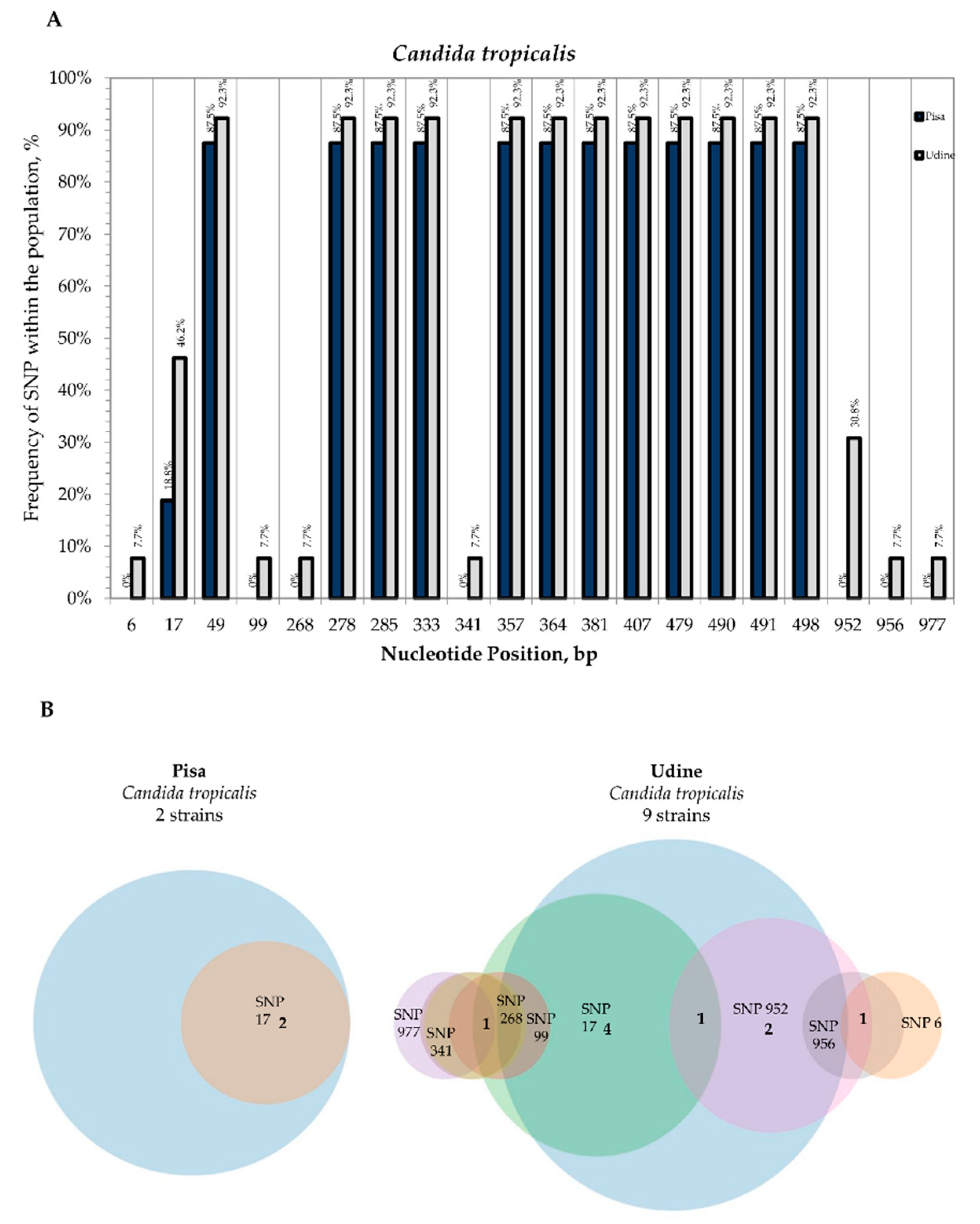
3.2. Internal Variability of Candida Species (SNP)
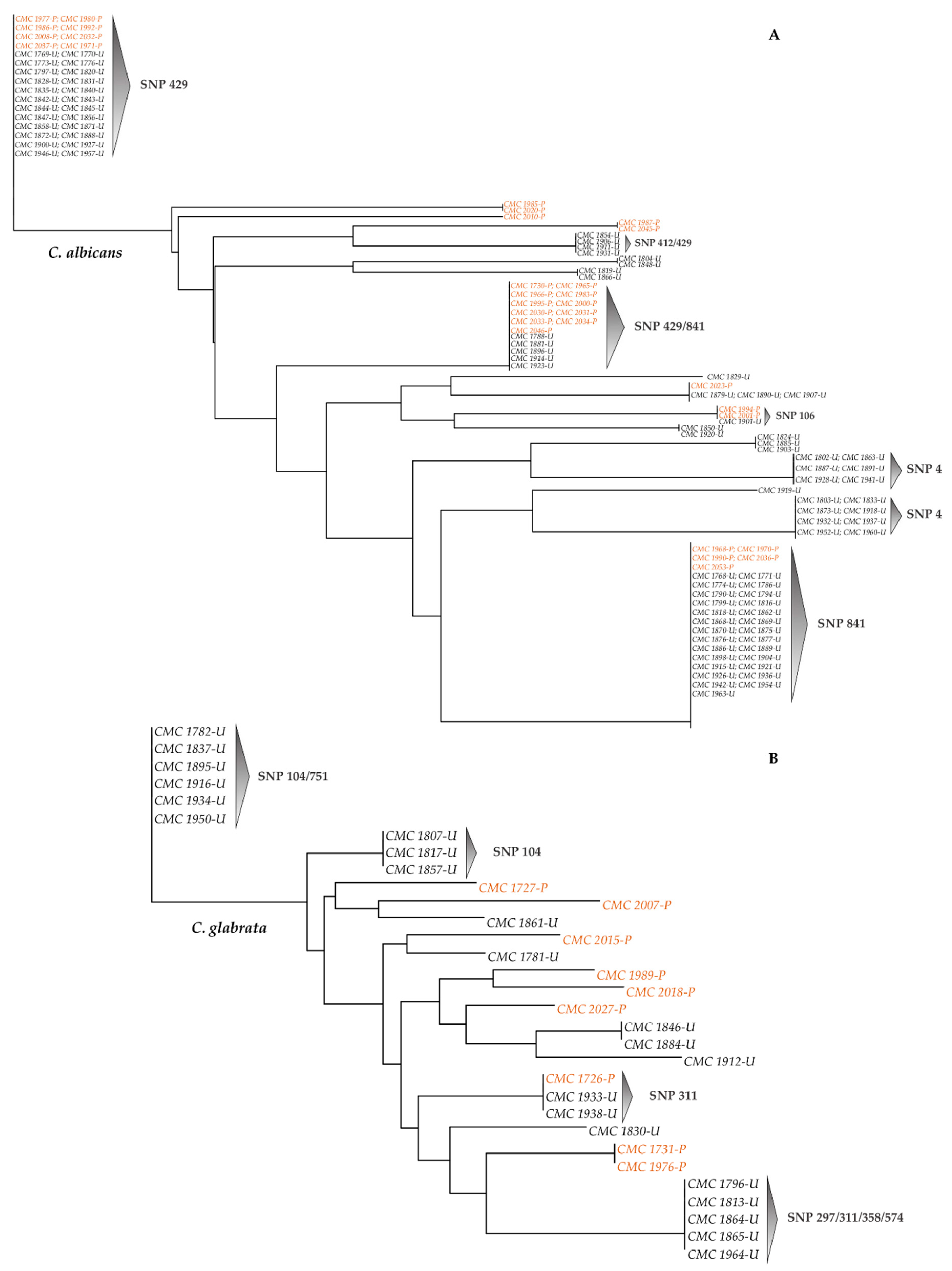
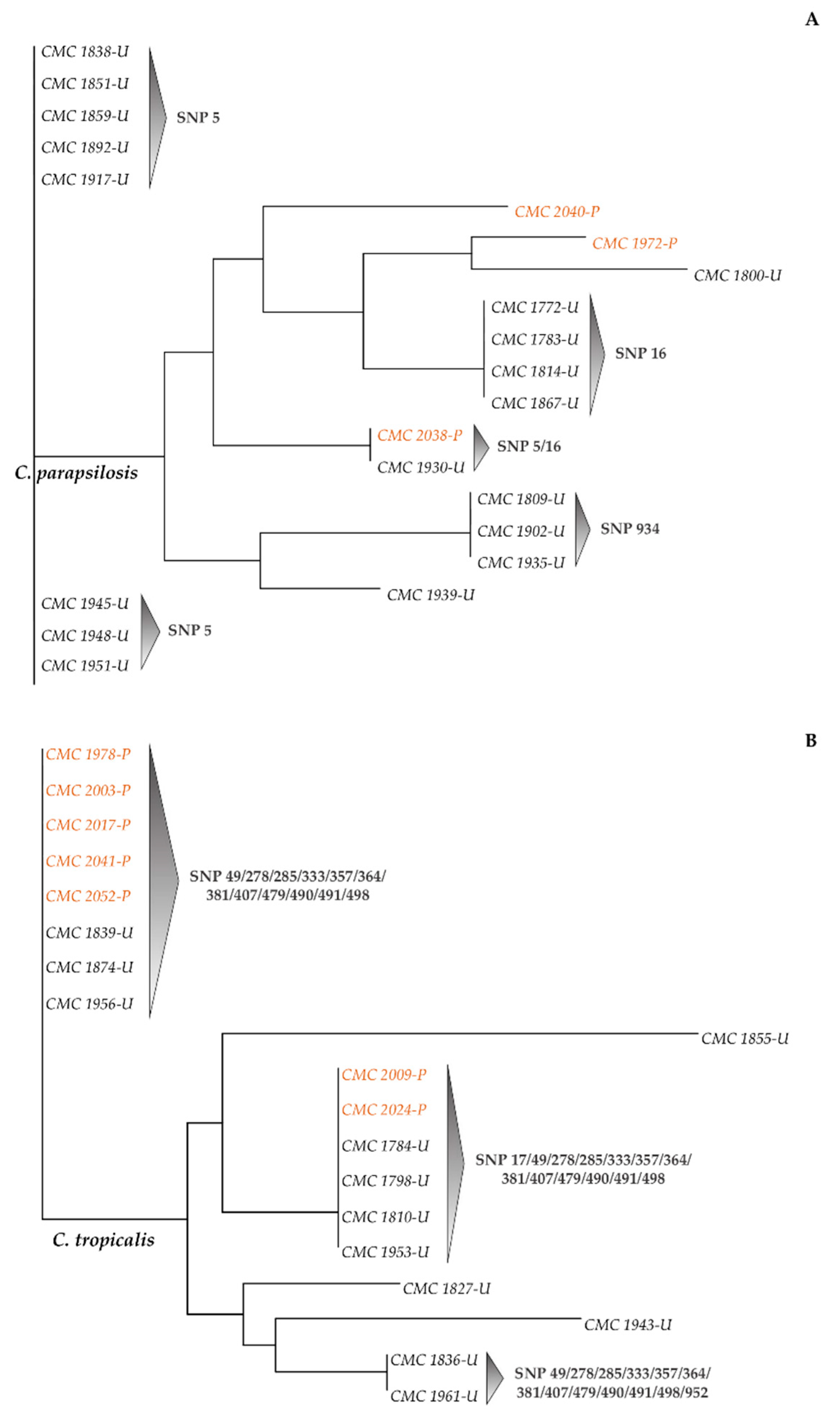
3.3. SNPs-Based Phylogenetic Analysis of Candida Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gutell, R.R.; Larsen, N.; Woese, C.R. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol. Mol. Biol. Rev. 1994, 58, 10–26. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Chapelle, S.; De Wachter, R. A quantitative map of nucleotide substitution rates in bacterial rRNA. Nucleic Acids Res. 1996, 24, 3381–3391. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.D.; Wensink, P.C.; Jordan, E.S. Xenopus laevis and Xenopus mulleri: The evolution of tandem gene. J. Mol. Biol. 1972, 63, 57–73. [Google Scholar] [CrossRef]
- Jeffreys, A. DNAsequence variants in the Gγ-,Aγ-, δ-and β-globin genes of man. Cell 1979, 18, 1–10. [Google Scholar] [CrossRef]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121. [Google Scholar]
- Rooney, A.P.; Ward, T.J. Evolution of a large ribosomal RNA multigene family in filamentous fungi: Birth and death of a concerted evolution paradigm. Proc. Natl. Acad. Sci. USA 2005, 102, 5084–5089. [Google Scholar] [CrossRef]
- Kurtzman, C.P. Identification of food and beverage spoilage yeasts from DNA sequence analyses. Int. J. Food Microbiol. 2015, 213, 71–78. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Leeuwenhoek 1998, 73, 331–371. [Google Scholar] [CrossRef]
- Čadež, N.; Raspor, P.; Turchetti, B.; Cardinali, G.; Ciafardini, G.; Veneziani, G.; Peter, G. Candida adriatica sp. nov. and Candida molendinolei sp. nov., two yeast species isolated from olive oil and its by-products. Int. J. Syst. Evol. Microbiol. 2012, 62, 2296–2302. [Google Scholar]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Irinyi, L.; Serena, C.; Garcia-Hermoso, D.; Arabatzis, M.; Desnos-Ollivier, M.; Vu, D.; Cardinali, G.; Arthur, I.; Normand, A.-C.; Giraldo, A. International Society of Human and Animal Mycology (ISHAM)-ITS reference DNA barcoding database—The quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 2015, 53, 313–337. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Robbertse, B.; Robert, V.; Vu, D.; Cardinali, G.; Irinyi, L.; Meyer, W.; Nilsson, R.H.; Hughes, K.; Miller, A.N.; et al. Finding needles in haystacks: Linking scientific names, reference specimens and molecular data for Fungi. Database J. Biol. Datrabase Curation 2014, 2014, bau061. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.R.; Roe, A.D.; Sperling, F.A. Multi-locus species delimitation in closely related animals and fungi: One marker is not enough. Mol. Ecol. 2012, 21, 4422–4436. [Google Scholar] [CrossRef] [PubMed]
- Meyer, W.; Irinyi, L.; Hoang, M.T.V.; Robert, V.; Garcia-Hermoso, D.; Desnos-Ollivier, M.; Yurayart, C.; Tsang, C.-C.; Lee, C.-Y.; Woo, P.C. Database establishment for the secondary fungal DNA barcode translational elongation factor 1α (TEF1α). Genome 2019, 62, 160–169. [Google Scholar] [CrossRef]
- Vu, D.; Groenewald, M.; De Vries, M.; Gehrmann, T.; Stielow, B.; Eberhardt, U.; Al-Hatmi, A.; Groenewald, J.; Cardinali, G.; Houbraken, J. Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud. Mycol. 2019, 92, 135–154. [Google Scholar] [CrossRef]
- Colabella, C.; Corte, L.; Roscini, L.; Bassetti, M.; Tascini, C.; Mellor, J.C.; Meyer, W.; Robert, V.; Vu, D.; Cardinali, G. NGS barcode sequencing in taxonomy and diagnostics, an application in “Candida” pathogenic yeasts with a metagenomic perspective. IMA Fungus 2018, 9, 91–105. [Google Scholar] [CrossRef]
- Sipiczki, M.; Horvath, E.; Pfliegler, W.P. Birth-and-death evolution and reticulation of ITS segments of Metschnikowia andauensis and Metschnikowia fructicola rDNA repeats. Front. Microbiol. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Stadler, M.; Lambert, C.; Wibberg, D.; Kalinowski, J.; Cox, R.J.; Kolařík, M.; Kuhnert, E. Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol. Prog. 2020, 19, 235–245. [Google Scholar] [CrossRef]
- Wu, Z.-W.; Wang, Q.-M.; Liu, X.-Z.; Bai, F.-Y. Intragenomic polymorphism and intergenomic recombination in the ribosomal RNA genes of strains belonging to a yeast species. Pichia Membr. Mycol. 2016, 7, 102–111. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 1–32. [Google Scholar] [CrossRef]
- Roscini, L.; Tristezza, M.; Corte, L.; Colabella, C.; Perrotta, C.; Rampino, P.; Robert, V.; Vu, D.; Cardinali, G.; Grieco, F. Early Ongoing Speciation of Ogataea uvarum sp. Nov. within the Grape Ecosystem Revealed by the Internal Variability Among the rDNA Operon Repeats. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Simon, U.K.; Weiß, M. Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol. Biol. Evol. 2008, 25, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Ganley, A.R.; Kobayashi, T. Highly efficient concerted evolution in the ribosomal DNA repeats: Total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 2007, 17, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Torres-Machorro, A.L.; Hernández, R.; Cevallos, A.M.; López-Villaseñor, I. Ribosomal RNA genes in eukaryotic microorganisms: Witnesses of phylogeny? FEMS Microbiol. Rev. 2010, 34, 59–86. [Google Scholar] [CrossRef] [PubMed]
- West, C.; James, S.A.; Davey, R.P.; Dicks, J.; Roberts, I.N. Ribosomal DNA sequence heterogeneity reflects intraspecies phylogenies and predicts genome structure in two contrasting yeast species. Syst. Biol. 2014, 63, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Corte, L.; Roscini, L.; Colabella, C.; Tascini, C.; Leonildi, A.; Sozio, E.; Menichetti, F.; Merelli, M.; Scarparo, C.; Meyer, W.; et al. Exploring ecological modelling to investigate factors governing the colonization success in nosocomial environment of Candida albicans and other pathogenic yeasts. Sci. Rep. 2016, 6, 26860. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Alami, M.M.; Xue, J.; Ma, Y.; Zhu, D.; Abbas, A.; Gong, Z.; Wang, X. Structure, Function, Diversity, and Composition of Fungal Communities in Rhizospheric Soil of Coptis chinensis Franch under a Successive Cropping System. Plants 2020, 9, 244. [Google Scholar] [CrossRef]
- Dresch, P.; Falbesoner, J.; Ennemoser, C.; Hittorf, M.; Kuhnert, R.; Peintner, U. Emerging from the ice-fungal communities are diverse and dynamic in earliest soil developmental stages of a receding glacier. Environ. Microbiol. 2019, 21, 1864–1880. [Google Scholar] [CrossRef]
- Kettner, M.T.; Rojas-Jimenez, K.; Oberbeckmann, S.; Labrenz, M.; Grossart, H.P. Microplastics alter composition of fungal communities in aquatic ecosystems. Environ. Microbiol. 2017, 19, 4447–4459. [Google Scholar]
- Schöler, A.; Jacquiod, S.; Vestergaard, G.; Schulz, S.; Schloter, M. Analysis of Soil Microbial Communities Based on Amplicon Sequencing of Marker Genes; Springer: Berlin, Germany, 2017. [Google Scholar]
- Pauvert, C.; Buée, M.; Laval, V.; Edel-Hermann, V.; Fauchery, L.; Gautier, A.; Lesur, I.; Vallance, J.; Vacher, C. Bioinformatics matters: The accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol. 2019, 41, 23–33. [Google Scholar] [CrossRef]
- Vesty, A.; Biswas, K.; Taylor, M.W.; Gear, K.; Douglas, R.G. Evaluating the impact of DNA extraction method on the representation of human oral bacterial and fungal communities. PLoS ONE 2017, 12, e0169877. [Google Scholar] [CrossRef] [PubMed]
- Chin, V.K.; Lee, T.Y.; Rusliza, B.; Chong, P.P. Dissecting Candida albicans infection from the perspective of C. albicans virulence and omics approaches on host–pathogen interaction: A review. Int. J. Mol. Sci. 2016, 17, 1643. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Lionakis, M.S.; Arendrup, M.C.; Ostrosky-Zeichner, L.; Kullberg, B.J. Invasive candidiasis. Nat. Rev. Dis. Primers 2018, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R.; Sherry, L.; Nile, C.J.; Sherriff, A.; Johnson, E.; Hanson, M.; Williams, C.; Munro, C.; Jones, B.; Ramage, G. Biofilm formation is a risk factor for mortality in patients with Candida albicans bloodstream infection—Scotland, 2012–2013. Clin. Microbiol. Infect. 2016, 22, 87–93. [Google Scholar] [CrossRef]
- Vu, D.; Groenewald, M.; Szöke, S.; Cardinali, G.; Eberhardt, U.; Stielow, B.; de Vries, M.; Verkley, G.J.M.; Crous, P.W.; Boekhout, T.; et al. DNA barcoding analysis of more than 9000 yeast isolates contributes to quantitative thresholds for yeast species and genera delimitation. Stud. Mycol. 2016, 85, 91–105. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Yurkov, A.; Guerreiro, M.A.; Sharma, L.; Carvalho, C.; Fonseca, Á. Multigene assessment of the species boundaries and sexual status of the basidiomycetous yeasts Cryptococcus flavescens and C. terrestris (Tremellales). PLoS ONE 2015, 10, e0120400. [Google Scholar]
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; de Montigny, J.; Marck, C.; Neuveglise, C.; Talla, E.; et al. Genome evolution in yeasts. Nature 2004, 430, 35–44. [Google Scholar] [CrossRef]
- Maleszka, R.; Clark-Walker, G. Yeasts have a four-fold variation in ribosomal DNA copy number. Yeast 1993, 9, 53–58. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colabella, C.; Casagrande Pierantoni, D.; Corte, L.; Roscini, L.; Conti, A.; Bassetti, M.; Tascini, C.; Robert, V.; Cardinali, G. Single Strain High-Depth NGS Reveals High rDNA (ITS-LSU) Variability in the Four Prevalent Pathogenic Species of the Genus Candida. Microorganisms 2021, 9, 302. https://doi.org/10.3390/microorganisms9020302
Colabella C, Casagrande Pierantoni D, Corte L, Roscini L, Conti A, Bassetti M, Tascini C, Robert V, Cardinali G. Single Strain High-Depth NGS Reveals High rDNA (ITS-LSU) Variability in the Four Prevalent Pathogenic Species of the Genus Candida. Microorganisms. 2021; 9(2):302. https://doi.org/10.3390/microorganisms9020302
Chicago/Turabian StyleColabella, Claudia, Debora Casagrande Pierantoni, Laura Corte, Luca Roscini, Angela Conti, Matteo Bassetti, Carlo Tascini, Vincent Robert, and Gianluigi Cardinali. 2021. "Single Strain High-Depth NGS Reveals High rDNA (ITS-LSU) Variability in the Four Prevalent Pathogenic Species of the Genus Candida" Microorganisms 9, no. 2: 302. https://doi.org/10.3390/microorganisms9020302
APA StyleColabella, C., Casagrande Pierantoni, D., Corte, L., Roscini, L., Conti, A., Bassetti, M., Tascini, C., Robert, V., & Cardinali, G. (2021). Single Strain High-Depth NGS Reveals High rDNA (ITS-LSU) Variability in the Four Prevalent Pathogenic Species of the Genus Candida. Microorganisms, 9(2), 302. https://doi.org/10.3390/microorganisms9020302