
Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Genomes
2.2. Cloning of Strain w15 16S rRNA Genes and Sequencing
2.3. Detection of 16S rRNA Gene Types—And Their Ratio—In Transcriptomes
2.4. Phylogenetic Analyses
2.5. Comparative Genome Analyses
2.6. Genome-Based Taxonomic Analyses
3. Results
3.1. General Properties of the Genomes of Strains Assigned or Related to S. multivorum and S. siyangense
3.2. Unraveling the 16S rRNA Gene Features of Strain w15 and Other Strains
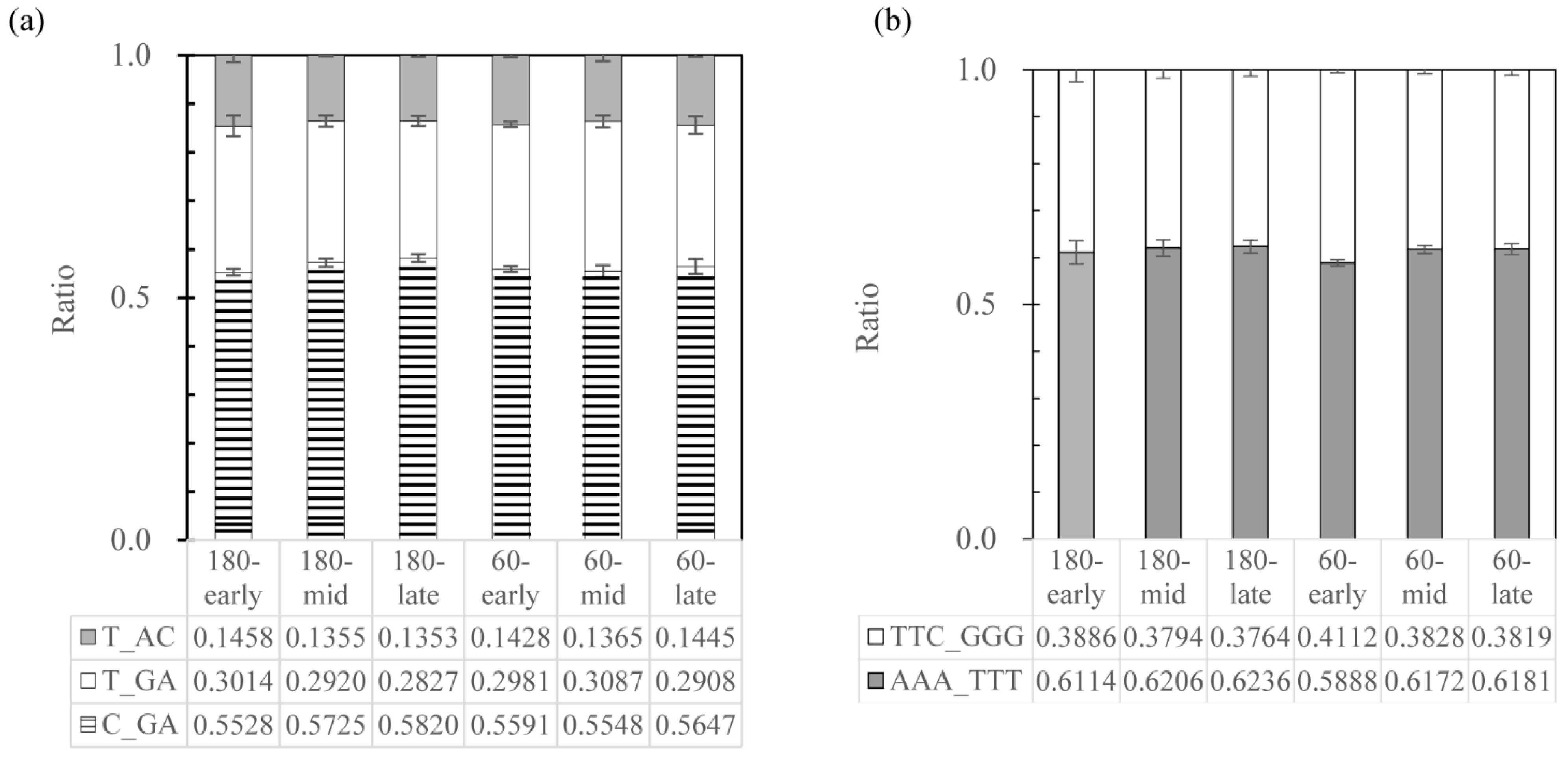
3.2.1. Small Regions in the 16S rRNA Gene V2 Region Drive the Microheterogeneity
3.2.2. Exploring the 16S rRNA Operons in Strain w15 and Related Strains
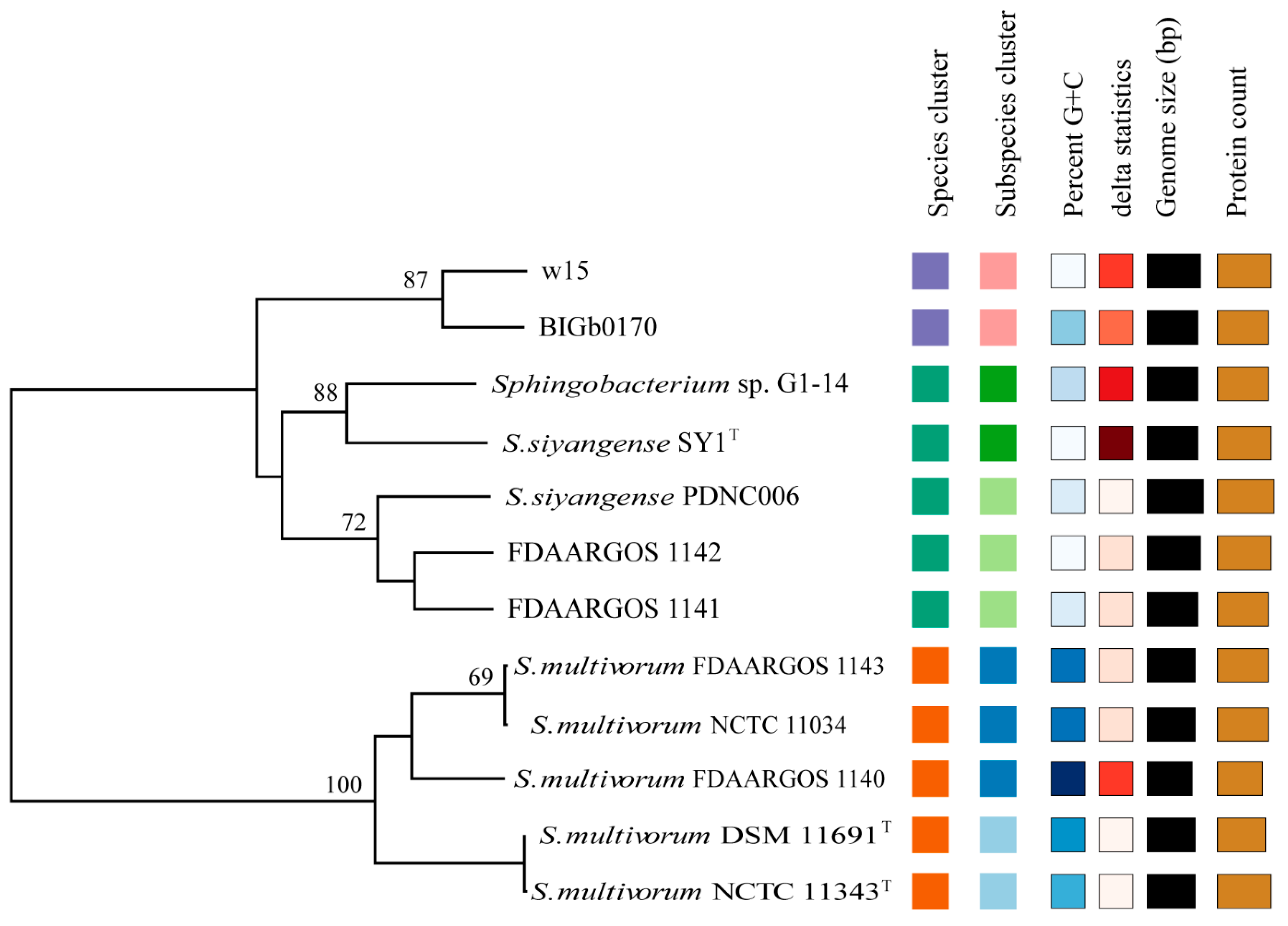
3.3. Phylogenetic Analyses
3.4. Average Nucleotide Identity (ANI) and Tetranucleotide Frequency (TETRA) Analyses
3.4.1. Taxonomic Placement of FDAARGOS 1141 and FDAARGOS 1142
3.4.2. Proposal of a New Species Based on Strains w15 and BIGb0170
3.5. Genome-Based Taxonomic Analyses
3.6. Examination of Strain Origins and Ecophysiological Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cortes-Tolalpa, L.; Jiménez, D.J.; de Lima Brossi, M.J.; Salles, J.F.; van Elsas, J.D. Different inocula produce distinctive microbial consortia with similar lignocellulose degradation capacity. Appl. Microbiol. Biotechnol. 2016, 100, 7713–7725. [Google Scholar] [CrossRef]
- Jiménez, D.J.; Maruthamuthu, M.; van Elsas, J.D. Metasecretome analysis of a lignocellulolytic microbial consortium grown on wheat straw, xylan and xylose. Biotechnol. Biofuels 2015, 8, 199. [Google Scholar] [CrossRef]
- Cortes-Tolalpa, L.; Salles, J.F.; van Elsas, J.D. Bacterial synergism in lignocellulose biomass degradation–complementary roles of degraders as influenced by complexity of the carbon source. Front. Microbiol. 2017, 8, 1628. [Google Scholar] [CrossRef]
- Ventorino, V.; Aliberti, A.; Faraco, V.; Robertiello, A.; Giacobbe, S.; Ercolini, D.; Amore, A.; Fagnano, M.; Pepe, O. Exploring the microbiota dynamics related to vegetable biomasses degradation and study of lignocellulose-degrading bacteria for industrial biotechnological application. Sci. Rep. 2015, 5, 8161. [Google Scholar] [CrossRef] [PubMed]
- Photphisutthiphong, Y.; Vatanyoopaisarn, S. Dyadobacter and Sphingobacterium isolated from herbivore manure in Thailand and their cellulolytic activity in various organic waste substrates. Agric. Nat. Resour. 2019, 53, 89–98. [Google Scholar] [CrossRef]
- Kitamikado, M.; Ito, M. Isolation of keratanase-producing bacteria from natural habitats. J. Fact. Agric. Kyushu Univ. 1979, 24, 101–112. [Google Scholar] [CrossRef]
- Malfliet, S.; Justé, A.; Crauwels, S.; Willems, K.; De Cooman, L.; Lievens, B.; Aerts, G. Assessing the xylanolytic bacterial diversity during the malting process. Food Microbiol. 2013, 36, 406–415. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Tian, C.; Xu, J.; Dong, F.; Liu, X.; Wu, X.; Zheng, Y. Characterization of hexaconazole-degrading strain Sphingobacterium multivorum and analysis of transcriptome for biodegradation mechanism. Sci. Total Environ. 2020, 722, 137171. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.A.; Mohamedelhassan, E.E.; Yanful, E.K.; Weselowski, B.; Yuan, Z.C. Isolation and characterization of novel bacterial strains for integrated solar-bioelectrokinetic of soil contaminated with heavy petroleum hydrocarbons. Chemosphere 2019, 237, 124514. [Google Scholar] [CrossRef]
- Cheng, J.; Sun, Z.; Yu, Y.; Li, X.; Li, T. Effects of modified carbon black nanoparticles on plant-microbe remediation of petroleum and heavy metal co-contaminated soils. Int. J. Phytoremed. 2019, 21, 634–642. [Google Scholar] [CrossRef]
- Balu, S.; Bhunia, S.; Gachhui, R.; Mukherjee, J. Assessment of polycyclic aromatic hydrocarbon contamination in the Sundarbans, the world’s largest tidal mangrove forest and indigenous microbial mixed biofilm-based removal of the contaminants. Environ. Pollut. 2020, 266, 115270. [Google Scholar] [CrossRef]
- Eke, P.; Kumar, A.; Sahu, K.P.; Wakam, L.N.; Sheoran, N.; Ashajyothi, M.; Patel, A.; Fekam, F.B. Endophytic bacteria of desert cactus (Euphorbia trigonas Mill) confer drought tolerance and induce growth promotion in tomato (Solanum lycopersicum L.). Microbiol. Res. 2019, 228, 126302. [Google Scholar] [CrossRef]
- Holmes, B.; Owen, R.J.; Weaver, R.E. Flavobacterium multivorum, a new species isolated from human clinical specimens and previously known as group IIk, biotype 2. Int. J. Syst. Bacteriol. 1981, 31, 21–34. [Google Scholar] [CrossRef]
- Abro, A.H.; Rahimi Shahmirzadi, M.R.; Jasim, L.M.; Badreddine, S.; Al Deesi, Z. Sphingobacterium multivorum Bacteremia and Acute Meningitis in an immunocompetent adult patient: A case report. Iran. Red Crescent Med. J. 2016, 18, e38750. [Google Scholar] [CrossRef][Green Version]
- Konala, V.M.; Naramala, S.; Bose, S.; Gayam, V.; Madhira, B.R.; Adapa, S. Bacteremia secondary to uncommon gram-negative Bacilli transmitted from the canine in a patient with multiple Myeloma. J. Investig. Med. High Impact Case Rep. 2020, 8. [Google Scholar] [CrossRef]
- Lambiase, A.; Rossano, F.; Del Pezzo, M.; Raia, V.; Sepe, A.; de Gregorio, F.; Catania, M.R. Sphingobacterium respiratory tract infection in patients with cystic fibrosis. BMC Res. Notes 2009, 2, 262. [Google Scholar] [CrossRef]
- Cortes-Tolalpa, L.; Wang, Y.; Salles, J.F.; van Elsas, J.D. Comparative genome analysis of the lignocellulose degrading bacteria Citrobacter freundii so4 and Sphingobacterium multivorum w15. Front. Microbiol. 2020, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, E.; Kaneko, T.; Yano, I.; Moss, C.W.; Miyoshi, N. Sphingobacterium gen. nov., Sphingobacterium spiritivorum comb. nov., Sphingobacterium multivorum comb. nov., Sphingobacterium mizutae sp. nov., and Flavobacterium indologenes sp. nov.: Glucose-Nonfermenting Gram-Negative Rods in CDC Groups IIK-2 and IIb. Int. J. Syst. Evol. Microbiol. 1983, 33, 580–598. [Google Scholar] [CrossRef]
- Kakumanu, M.L.; Marayati, B.F.; Wada-Katsumata, A.; Wasserberg, G.; Schal, C.; Apperson, C.S.; Ponnusamy, L. Sphingobacterium phlebotomi sp. nov., a new member of family Sphingobacteriaceae isolated from sand fly rearing substrate. Int. J. Syst. Evol. Microbiol. 2021, 71, 004809. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, H.; Zhang, C.X.; Yang, S.Y.; Liu, X.H.; Zhang, K.Y.; Lai, R. Sphingobacterium siyangense sp. nov., isolated from farm soil. Int. J. Syst. Evol. Microbiol. 2008, 58, 1458–1462. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar Chaudhary, D.; Kim, J. Sphingobacterium terrae sp. nov., isolated from oil-contaminated soil. Int. J. Syst. Evol. Microbiol. 2018, 68, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; Owen, R.J.; Hollis, D.G. Flavobacterium spiritittorum, a new species isolated from human clinical specimens. Int. J. Syst. Bacteriol. 1982, 32, 157–165. [Google Scholar] [CrossRef]
- Dirksen, P.; Assié, A.; Zimmermann, J.; Zhang, F.; Tietje, A.M.; Marsh, S.A.; Félix, M.A.; Shapira, M.; Kaleta, C.; Schulenburg, H.; et al. CeMbio—The Caenorhabditis elegans Microbiome Resource. G3 Genes Genomes Genet. 2020, 10, 3025–3039. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Pride, D.T.; Meinersmann, R.J.; Wassenaar, T.M.; Blaser, M.J. Evolutionary implications of microbial genome tetranucleotide frequency biases. Genome Res. 2003, 13, 145–158. [Google Scholar] [CrossRef]
- Grizard, S.; Dini-Andreote, F.; Tieleman, B.I.; Salles, J.F. Dynamics of bacterial and fungal communities associated with eggshells during incubation. Ecol. Evol. 2014, 4, 1140–1157. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Glöckner, F.O.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Gen. Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Gen. Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yokota, A. Proposals of Sphingobacterium faecium sp. nov., Sphingobacterium piscium sp. nov., Sphingobacterium heparinum comb. nov., Sphingobacterium thalpophilum comb. nov., and two genospecies of the genus Sphingobacterium, and synonymy of Flavobacterium yabuuchiae and Sphingobacterium spiritivorum. J. Gen. Appl. Microbiol. 1992, 38, 465–482. [Google Scholar] [CrossRef]
- Steyn, P.L.; Segers, P.; Vancanneyt, M.; Sandra, P.; Kersters, K.; Joubert, J.J. Classification of heparinolytic bacteria into a new genus, Pedobacter, comprising four species: Pedobacter heparinus comb. nov., Pedobacter piscium comb. nov., Pedobacter africanus sp. nov. and Pedobacter saltans sp. nov. Proposal of the family Sphingobacteriaceae fam. nov. Int. J. Sys. Bact. 1998, 48, 165–177. [Google Scholar] [CrossRef]
- Oyaizu, H.; Komagata, K. Chemotaxanomic and phenotypic characterization of the strains of species in the Flavobacterium-cytophaga complex. J. Gen. Appl. Microbiol. 1981, 27, 57–107. [Google Scholar] [CrossRef]
- Yin, P.; Liu, S.; Wu, X. Whole genome sequence of Sphingobacterium sp. G1-14, a strain with effective paichongding biodegradation. J. Biol. Life Sci. 2019, 10, 58. [Google Scholar] [CrossRef]
- Lee, D.; Hur, J.S.; Kahng, H.Y. Sphingobacterium cladoniae sp. nov., isolated from lichen, Cladonia sp., and emended description of Sphingobacterium siyangense. Int. J. Syst. Evol. Microbiol. 2013, 63, 755–760. [Google Scholar] [CrossRef]
- Tang, L.; Li, Y.; Deng, X.; Johnston, R.N.; Liu, G.R.; Liu, S.L. Defining natural species of bacteria: Clear-cut genomic boundaries revealed by a turning point in nucleotide sequence divergence. BMC Genom. 2013, 14, 489. [Google Scholar] [CrossRef]
- Bobay, L.M.; Ochman, H. The Evolution of Bacterial Genome Architecture. Front Genet. 2017, 30, 72. [Google Scholar] [CrossRef]
- Mira, A.; Ochman, H.; Moran, N.A. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001, 17, 589–596. [Google Scholar] [CrossRef]
- Condon, C.; Liveris, D.; Squires, C.; Schwartz, I.; Squires, C.L. rRNA operon multiplicity in Escherichia coli and the physiological implications of rrn inactivation. J. Bacteriol. 1995, 177, 4152–4156. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, B.S.; Schmidt, T.M. Life history implications of rRNA gene copy number in Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 6670–6677. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, P.M.; Noll, M.; Liesack, W. Phylogenetic identity, growth-response time and rRNA operon copy number of soil bacteria indicate different stages of community succession. Environ. Microbiol. 2007, 9, 2464–2474. [Google Scholar] [CrossRef]
- Roller, B.R.K.; Schmidt, T.M. The physiology and ecological implications of efficient growth. ISME J. 2015, 9, 1481–1487. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar] [CrossRef]
- Stevenson, B.S.; Schmidt, T.M. Growth rate-dependent expression of RNA from plasmid-borne rRNA operons in Escherichia coli. J. Bacteriol. 1998, 180, 1970–1972. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genome Size (Mb) | GC% | Total Genes | Protein Encoding Genes | Pseudo Genes | 5S rRNA | 16S rRNA | 23S rRNA | tRNA | RefSeq Assembly Accession | Gene Load (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| w15 | 6.68 | 39.8 | 5541 | 5426 | 40 | 7 | 6 a | 1 * | 63 | GCF_009660355.1 | 82.95 |
| BIGb0170 | 6.43 | 39.9 | 5348 | 5222 | 19 | 7 | 7 | 7 | 83 | GCF_014109745.1 | 83.17 |
| S.multivorum NCTC11343T | 5.98 | 40.0 | 5310 | 4685 | 517 | 7 | 7 | 8 | 83 | GCF_900457465.1 | 88.80 |
| S.multivorum NCTC11034 | 6.01 | 40.1 | 5207 | 5050 | 53 | 6 | 6 | 6 | 83 | GCF_900457115.1 | 86.64 |
| S.multivorum DSM11691T | 5.97 | 40.0 | 5126 | 4944 | 75 | 7 | 7 | 7 | 83 | GCF_016894225.1 | 85.86 |
| S.multivorum FDAARGOS 1140 | 5.66 | 40.1 | 4879 | 4718 | 50 | 7 | 7 | 7 | 87 | GCF_016726045.1 | 86.20 |
| S.multivorum FDAARGOS 1143 | 6.02 | 40.1 | 5221 | 5062 | 50 | 7 | 7 | 7 | 85 | GCF_016725865.1 | 86.73 |
| FDAARGOS 1141 | 6.29 | 39.8 | 5274 | 5156 | 11 | 7 | 7 | 7 | 83 | GCF_016725925.1 | 83.85 |
| FDAARGOS 1142 | 6.55 | 39.8 | 5527 | 5402 | 18 | 7 | 7 | 7 | 83 | GCF_016725465.1 | 84.38 |
| S. siyangense SY1T | 6.29 | 39.8 | 5482 | 5330 | 79 | 5 | – b | – | 65 | GCF_007830445.1 | 87.15 |
| S. siyangense PDNC006 | 6.83 | 39.8 | 5741 | 5607 | 27 | 7 | 7 | 7 | 83 | GCF_016919365.1 | 84.06 |
| Sphingobacterium sp. G1-14 | 6.33 | 39.8 | 5388 | 5235 | 46 | 7 | 7 | 7 | 83 | GCF_002196555.1 | 85.12 |
| Similarity to | |||||
|---|---|---|---|---|---|
| Strain | 16S rRNA Designation | Accession no. | Sequences at Position 186–210 | Canonical | Divergent |
| divergent (1/7) | UAUU01000005.1_rrna_60 | CAT--A-A-TTCTCCGGCATCGGAGTATT | 98.89 | 100.00 | |
| S. multivorum | UAUU01000002.1_rrna_32 | CATCAACAGTTCGCATG-TTC-G-GT-TG | 99.61 | 99.02 | |
| UAUU01000009.1_rrna_17 | CATCAACAGTTCGCATG-TTC-G-GT-TG | 99.93 | 98.96 | ||
| NCTC 11343T | UAUU01000009.1_rrna_23 | CATCAACAGTTCGCATG-TTC-G-GT-TG | 99.54 | 98.96 | |
| canonical (6/7) | UAUU01000011.1_rrna_80 | TATCAACAGTTCGCATG-TTC-G-GT-TG | 100.00 | 98.89 | |
| UAUU01000002.1_rrna_47 | CATCAACAGTTCGCATG-TTC-G-GT-TG | 99.74 | 98.76 | ||
| UAUU01000009.1_rrna_1 | CATCAACAGTTCGCATG-TTC-G-GT-TG | 99.77 | 98.63 | ||
| Ratio | Position 186 | Position 207–209 | Position 463–465—474–476 | Cloning No. (1367 bp) |
|---|---|---|---|---|
| 11/40 | C | G-A | AAA-TTT | 5, 9, 12, 15, 29, 30, 32, 33, 34, 38, 39 |
| 8/40 | T | G-A | AAA-TTT | 3, 4, 7, 10, 11, 21, 28, 35 |
| 3/40 | T | A-C | AAA-TTT | 8, 14, 16 |
| 11/40 | C | G-A | TTC-GGG | 1, 2, 18, 22, 24, 25, 26, 27, 31, 36, 37 |
| 2/40 | T | G-A | TTC-GGG | 20, 23 |
| 1/40 | T | A-C | TTC-GGG | 40 |
| V2 Region | V3 Region | Similarity to | ||||||
|---|---|---|---|---|---|---|---|---|
| Strain | 16S Designation | Accession number | 186–210 bp | 463–465/ 474–476 bp | w15 | NCTC Canonical | 11343T Divergent | SY1T |
| seq 1 (11/40) | PHGU01000070.1_rrna_66 | CATATCTGACCGGCATCGGTTGGAT | AAA/TTT | 100.00 | 98.69 | 98.89 | 99.06 | |
| seq 2 (8/40) | cloning 28 | TATATCTGACCGGCATCGGTTGGAT | AAA/TTT | 99,93 | 98.69 | 98.76 | 99.12 | |
| w15 | seq 3 (11/40) | cloning 36 | CATATCTGACCGGCATCGGTTGGAT | TTC/GGG | 99,49 | 98.10 | 98.32 | 98.83 |
| seq 4 (2/40) | cloning 23 | TATATCTGACCGGCATCGGTTGGAT | TTC/GGG | 99.34 | 98.10 | 98.17 | 98.68 | |
| seq 5 (1/40) | cloning 40 | TATATCTGACCGGCATCGGTTAGCT | TTC/GGG | 99.27 | 98.10 | 98.25 | 98.90 | |
| seq 2 (3/7) | CP058555.1_rrna_100 | TATATCTGACCGGCATCGGTTGGAT | TTC/GGG | 99.54 | 98.37 | 98.43 | 98.72 | |
| BIGb0170 | seq 1 (4/7) | CP058555.1_rrna_67 | CATATCTGACCGGCATCGGTTAGCT | TTC/GGG | 99.48 | 98.24 | 98.50 | 98.92 |
| S. multivorum | canonical (6/7) | UAUU01000011.1_rrna_80 | TATCAACAGTTC-GCAT-GTTCGGTTG | AAA/TTT | 98.69 | 100.00 | 98.89 | 98.32 |
| NCTC 11343T | divergent (1/7) | UAUU01000005.1_rrna_60 | CATAATTCTCCGGCATCGG-AGTATT | AAA/TTT | 98.89 | 98.89 | 100.00 | 98.86 |
| S. siyangense SY1T | EU046272.1 | CATATCTGACCGGCATCGGTTAGCT | AAG/TTC | 99.06 | 98.32 | 98.86 | 100.00 | |
| S. siyangense | (5/7) | CP070350.1_rrna_27 | CATATCTGACCGGCATCGGTTAGCT | AGA/TCT | 99.41 | 98.57 | 99.08 | 99.66 |
| PDNC006 | canonical (2/7) | CP070350.1_rrna_92 | TATCAACAGTTC-GCAT-GTTCAGTTG | AGA/TCT | 98.83 | 99.48 | 98.63 | 98.72 |
| seq 3 (1/7) | CP068224.1_rrna_6 | CATATCTGACCGGCATCGGTTGGAT | AGA/TCT | 99.67 | 98.76 | 98.95 | 99.40 | |
| FDAARGOS 1141 | seq 2 (2/7) | CP068224.1_rrna_91 | CATATGTGACCGGCATCGGTTGGAT | TTC/GGG | 99.28 | 98.50 | 98.76 | 98.99 |
| seq 1 (4/7) | CP068224.1_rrna_64 | CATATGTGACCGGCATCGGTTGGAT | TTC/GGG | 99.28 | 98.50 | 98.76 | 98.99 | |
| seq 2 (3/7) | CP068223.1_rrna_23 | CATATGTGACCGGCATCGGTTGGAT | AGA/TCT | 99.54 | 98.76 | 99.02 | 99.40 | |
| FDAARGOS 1142 | seq 1 (4/7) | CP068223.1_rrna_64 | CATATCTGACCGGCATCGGTTTGAT | TTC/GGG | 99.28 | 98.37 | 98.63 | 98.92 |
| W-like (1/7) | CP068089.1_rrna_11 | TATATCTGACCGGCATCGGTTAGCT | AAA/TTT | 99.22 | 99.02 | 99.15 | 99.06 | |
| S. multivorum | divergent (2/7) | CP068089.1_rrna_18 | CATAATTCTCTGGCATCGG-AGTATT | AAA/TTT | 98.76 | 98.83 | 99.87 | 98.72 |
| FDAARGOS 1140 | canonical (4/7) | CP068089.1_rrna_80 | TATCAACAGTTC-GCAT-GTTCGGTTG | AAA/TTT | 98.63 | 99.80 | 99.09 | 98.52 |
| S. multivorum | divergent (1/7) | CP068086.1_rrna_91 | CATAATTCTCTGGCATCGG-AGTATT | AAA/TTT | 98.82 | 99.02 | 99.67 | 98.52 |
| FDAARGOS 1143 | canonical (6/7) | CP068086.1_rrna_98 | CATCAACAGTTC-GCAT-GTTCGGTTG | AAA/TTT | 98.69 | 99.67 | 98.83 | 98.26 |
| Strain | Conclusion | Identification Result | Species Cluster | Subspecies Cluster |
|---|---|---|---|---|
| w15 | proposed new species | Sphingobacterium paramultivorum | 3 | 2 |
| BIGb0170 | proposed new species | Sphingobacterium paramultivorum | 3 | 2 |
| S. multivorum NCTC 11343T | belongs to known species | Sphingobacterium multivorum | 2 | 1 |
| S. multivorum DSM 11691T | belongs to known species | Sphingobacterium multivorum | 2 | 1 |
| S. multivorum NCTC 11034 | belongs to known species | Sphingobacterium multivorum | 2 | 0 |
| S. multivorum FDAARGOS 1140 | belongs to known species | Sphingobacterium multivorum | 2 | 0 |
| S. multivorum FDAARGOS 1143 | belongs to known species | Sphingobacterium multivorum | 2 | 0 |
| S. siyangense SY1T | belongs to known species | Sphingobacterium siyangense | 1 | 4 |
| Sphingobacterium sp. G1-14 | belongs to known species | Sphingobacterium siyangense | 1 | 4 |
| FDAARGOS 1141 | belongs to known species | Sphingobacterium siyangense | 1 | 3 |
| FDAARGOS 1142 | belongs to known species | Sphingobacterium siyangense | 1 | 3 |
| S. siyangense PDNC006 | belongs to known species | Sphingobacterium siyangense | 1 | 3 |
| Strains | Sp. w15 | Sp. BIGb 0170 | Ss. SY1T | Sm. NCTC 11343T | Ss. FDAARGOS 1141 | Ss. FDAARGOS 1142 | Sm. FDAARGOS 1140 | Sm. NCTC 11034 | Ssp. G1-14 | Ss. PDNC 006 |
|---|---|---|---|---|---|---|---|---|---|---|
| Isolation Source | Decaying Wood | Rotting Apple h | Farm Soil | Spleen | Soil | Activated Sludge | Succinoglycan | Blood | Soil | Plastic Debris |
| Growth at: | ||||||||||
| 4 °C | − | + | − | − | − | − | − | |||
| 42 °C | − | + | − | − | − | − | − | |||
| pH 4.0 | − | + | − | − | ||||||
| NaCl 5% | − | − | − | + | − | − | − | |||
| Tween 80 | − | − | − | + | + | + | + | |||
| L-arginine | − | − | + | − | ||||||
| D-sorbitol | − | + | − | |||||||
| L-sorbose | − | + | − | |||||||
| D-mannitol | − | − | + | − | ||||||
| Xylitol | − | + | − | |||||||
| Adonitol | − | + | − | |||||||
| Glycerol | − | + | − | |||||||
| D-mannose | + | + | − | − | ||||||
| L-rhamnose | + | + | − | |||||||
| α-Methyl-D-glucoside | + | + | + | |||||||
| L-asparagine | − | − | ||||||||
| L-phenylalanine | − | − | ||||||||
| i-erythritol | − | − | + | |||||||
| D-galacturonic acid | − | − | ||||||||
| D-galactonic acid lactone | − | − | ||||||||
| Putrescine | − | − | ||||||||
| L-threonine | − | − | v | |||||||
| Glycogen | − | − | v | |||||||
| Itaconic acid | − | − | ||||||||
| Pectin | + | + | − | |||||||
| Dextrin | + | − | + | |||||||
| Inulin | + | + | v | + | + | |||||
| Key enzymes produced: | ||||||||||
| a | b | c | c | c | d | |||||
| References | [17] | [23] | [20] | [13,18,40,41] | [6,40] | [41,42] | [41,42] | [13,18] | [43] | |
| Traits shared by all three clusters: | ||||||||||
| Gram-negative, aerobic growth, no motility, rod shaped cells | ||||||||||
| Carbon sources used by all three clusters: D-xylose, maltose, D-melibiose, D-fructose, D-glucose, sucrose, D-galactose, trehalose, lactose, cellobiose, melezitose, salicin, N-acetyl-D-glucosamine | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Brons, J.K.; van Elsas, J.D. Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum. Microorganisms 2021, 9, 2057. https://doi.org/10.3390/microorganisms9102057
Wang Y, Brons JK, van Elsas JD. Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum. Microorganisms. 2021; 9(10):2057. https://doi.org/10.3390/microorganisms9102057
Chicago/Turabian StyleWang, Yanfang, Jolanda K. Brons, and Jan Dirk van Elsas. 2021. "Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum" Microorganisms 9, no. 10: 2057. https://doi.org/10.3390/microorganisms9102057
APA StyleWang, Y., Brons, J. K., & van Elsas, J. D. (2021). Considerations on the Identity and Diversity of Organisms Affiliated with Sphingobacterium multivorum—Proposal for a New Species, Sphingobacterium paramultivorum. Microorganisms, 9(10), 2057. https://doi.org/10.3390/microorganisms9102057