
Novakomyces olei sp. nov., the First Member of a Novel Taphrinomycotina Lineage



Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Characterization
2.2. DNA Amplification, Sequencing, and Phylogenetic Analysis
2.3. Genome Sequencing, Assembly, and Annotation
2.4. Phylogenomic Analyses
2.5. Identification of Missing Genes Their Gene Ontology (GO) Enrichment
2.6. Genome Searches for Genes Involved in Assimilation of Sugars
3. Results and Discussion
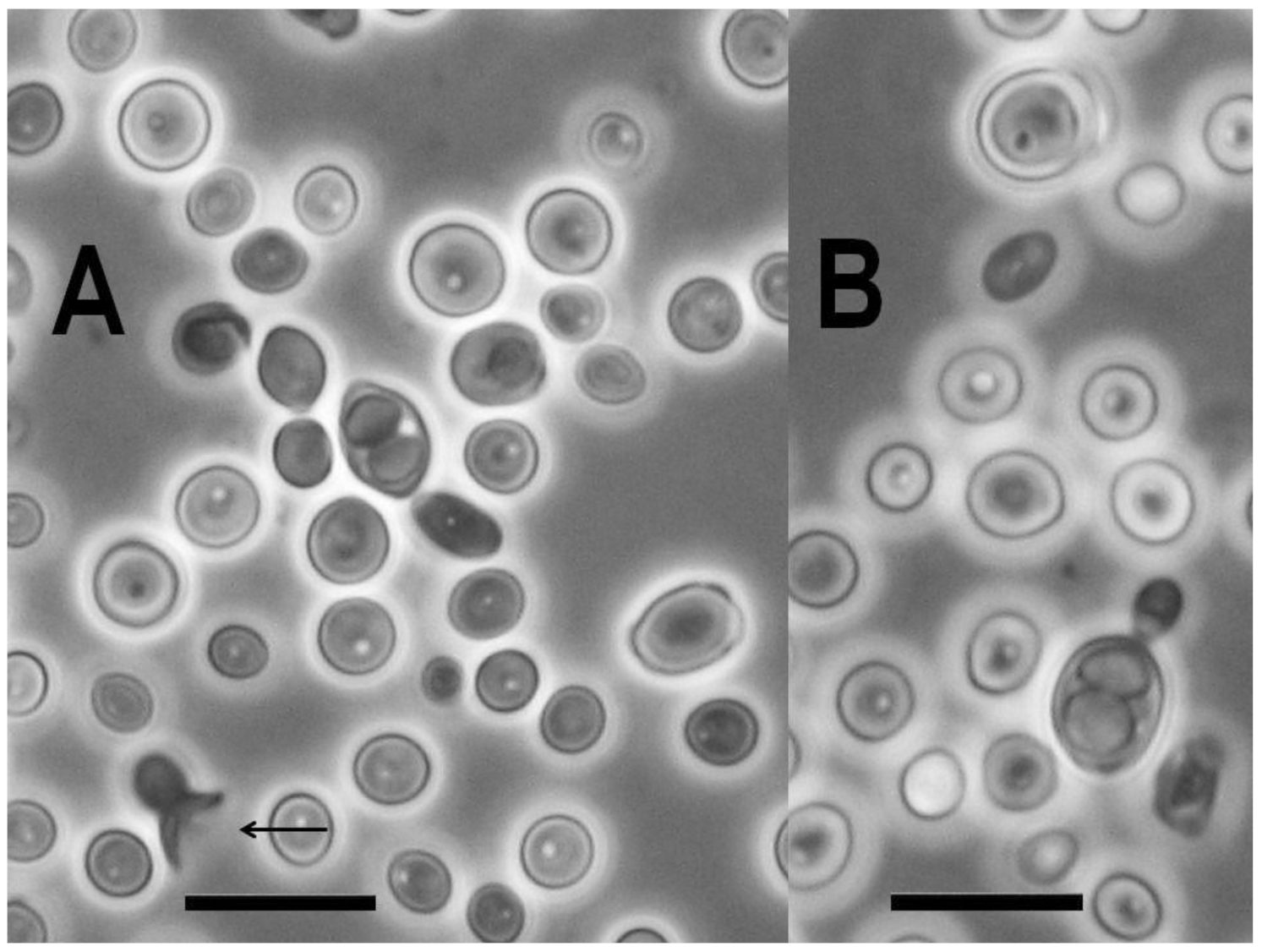
3.1. Isolation and Occurrence
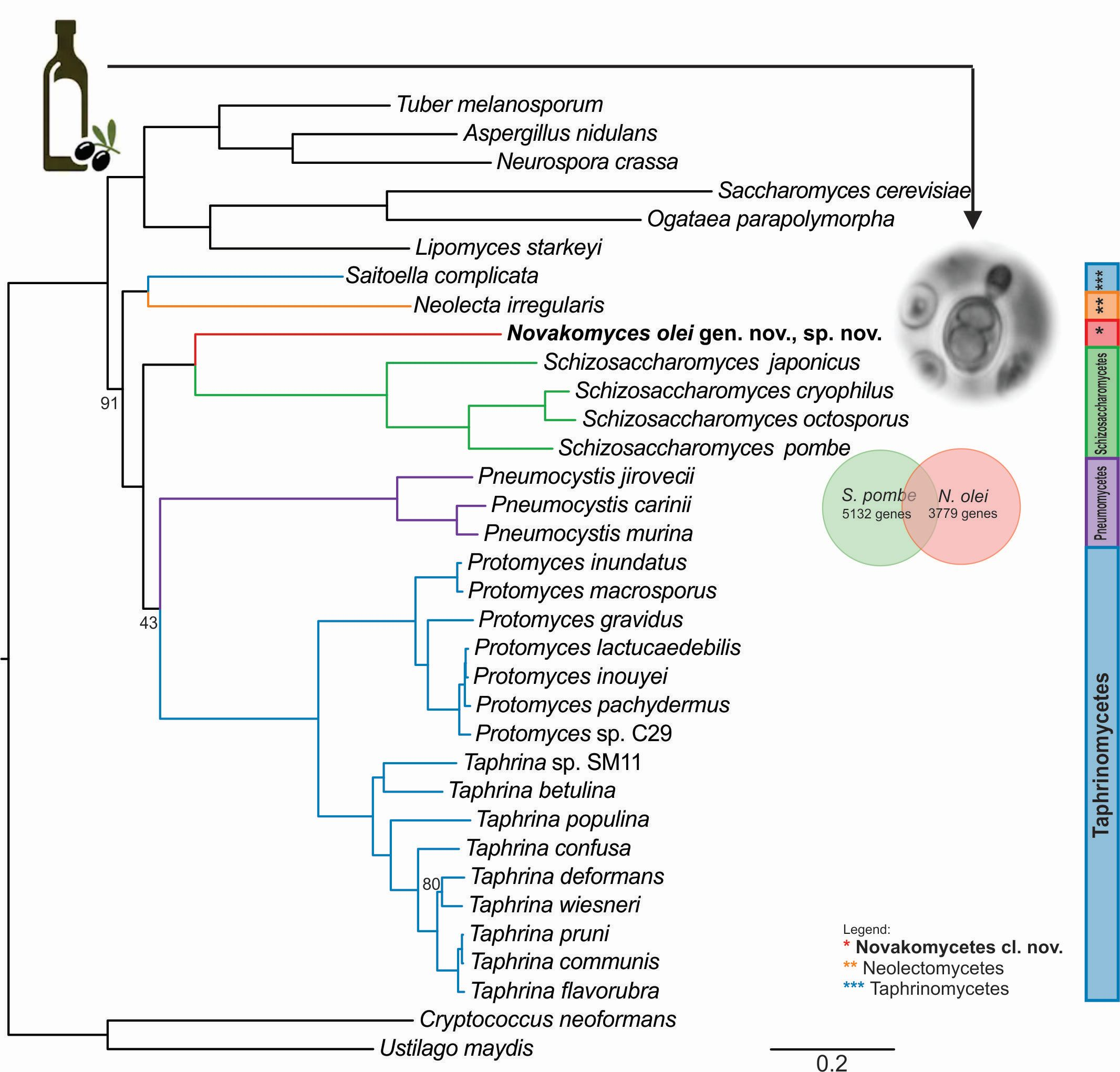
3.2. Ribosomal Gene Sequence Comparisons and Phylogenetic Placement Based on LSU Sequences
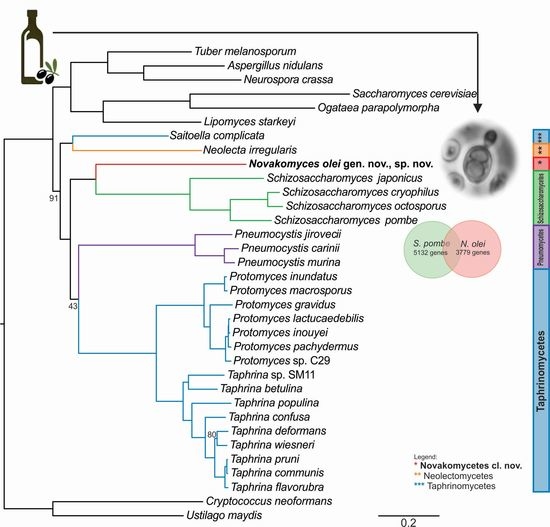
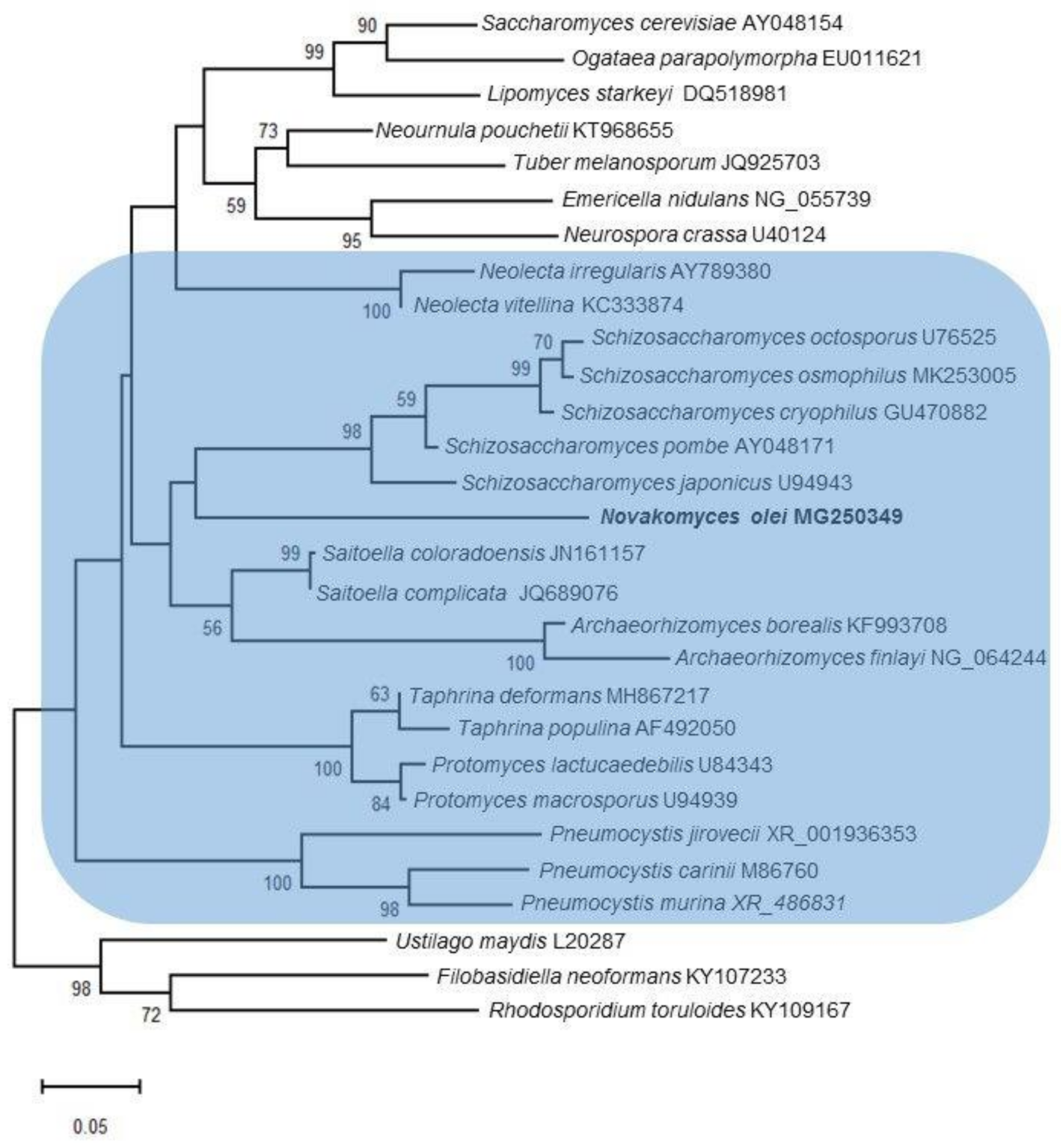
3.3. Phylogenomic Placement of the Novel Taxon
3.4. Content of Novakozyma olei Genome
3.5. Functional Analysis of Genes Missing in the Genome of Novakomyces olei
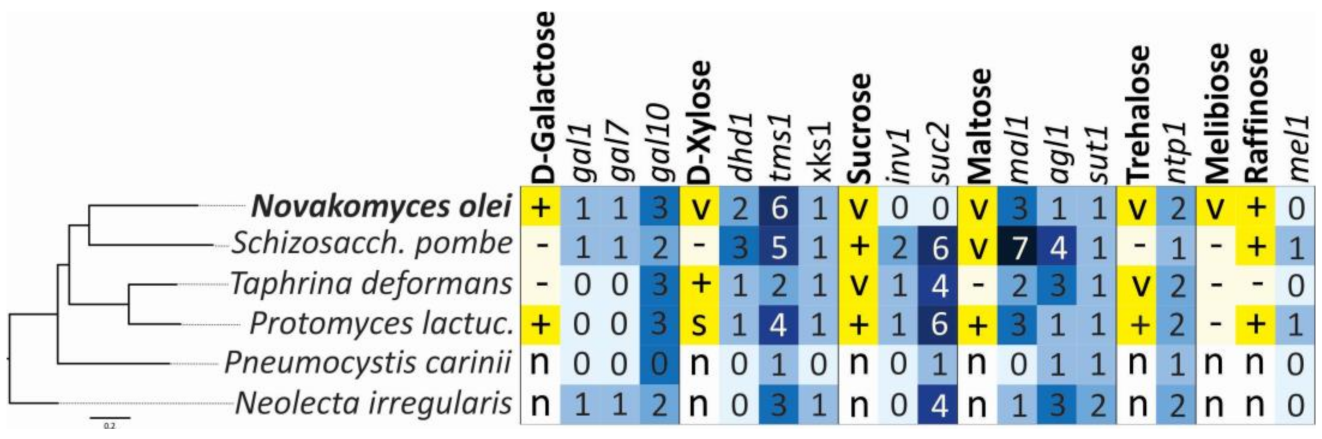
3.6. Phenotypic Characters and Their Correlation with Genome Content
3.7. Taxonomy
3.7.1. Novakomycetes Dlauchy, Péter & Čadež cl. nov.
3.7.2. Novakomycetales Dlauchy, Péter & Čadež ord. nov.
3.7.3. Novakomycetaceae Dlauchy, Péter & Čadež fam. nov.
3.7.4. Novakomyces Dlauchy, Péter & Čadež gen. nov.
3.7.5. Novakomyces olei Dlauchy, Péter & Čadež sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eriksson, O.E.; Winka, K. Supraordinal taxa of Ascomycota. Myconet 1997, 1, 1–16. [Google Scholar]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lücking, R.; et al. A higher-level phylogenetic classification of the Fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, J.; Hosaka, K.; Suh, S.-O. Early diverging Ascomycota: Phylogenetic divergence and related evolutionary enigmas. Mycologia 2006, 98, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Spatafora, J.W.; Aime, M.C.; Grigoriev, I.V.; Martin, F.; Stajich, J.E.; Blackwell, M. The Fungal Tree of Life: From Molecular Systematics to Genome-Scale Phylogenies. Microbiol. Spectr. 2017, 5, 3–34. [Google Scholar] [CrossRef]
- Nishida, H.; Sugiyama, J. Archiascomycetes: Detection of a major new lineage within the Ascomycota. Mycoscience 1994, 35, 361–366. [Google Scholar] [CrossRef]
- Crous, P.W.; Gams, W.; Stalpers, J.A.; Robert, V.; Stegehuis, G. MycoBank: An online initiative to launch mycology into the 21st century. Stud. Mycol. 2004, 50, 19–22. [Google Scholar]
- MYCOBANK Database. Available online: https://www.mycobank.org/ (accessed on 9 December 2020).
- Liu, Y.; Leigh, J.W.; Brinkmann, H.; Cushion, M.T.; Rodriguez-Ezpeleta, N.; Philippe, H.; Lang, B.F. Phylogenomic Analyses Support the Monophyly of Taphrinomycotina, including Schizosaccharomyces Fission Yeasts. Mol. Biol. Evol. 2009, 26, 27–34. [Google Scholar] [CrossRef]
- Riley, R.; Haridas, S.; Wolfe, K.H.; Lopes, M.R.; Hittinger, C.T.; Göker, M.; Salamov, A.A.; Wisecaver, J.H.; Long, T.M.; Calvey, C.H.; et al. Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA 2016, 113, 9882–9887. [Google Scholar] [CrossRef]
- Choi, J.; Kim, S.-H. A genome Tree of Life for the Fungi kingdom. Proc. Natl. Acad. Sci. USA 2017, 114, 9391–9396. [Google Scholar] [CrossRef]
- Rajeh, A.; Lv, J.; Lin, Z.G. Heterogeneous rates of genome rearrangement contributed to the disparity of species richness in Ascomycota. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Shen, X.X.; Steenwyk, J.L.; LaBella, A.L.; Opulente, D.A.; Zhou, X.; Kominek, J.; Li, Y.; Groenwald, M.; Hittinger, C.T.; Rokas, A. Genome-scale phylogeny and con-trasting modes of genome evolution in the fungal phylum Ascomycota. Sci. Adv. 2020, 6, eabd0079. [Google Scholar] [CrossRef] [PubMed]
- Vaughan-Martini, A.; Martini, A. Schizosaccharomyces Lindner (1983). In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 779–784. [Google Scholar] [CrossRef]
- Cushion, M.T.; Keely, S.P. Pneumocystis Delanoe & Delanoe (1912). In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 709–717. [Google Scholar] [CrossRef]
- Fonseca, A.; Rodrigues, M. Taphrina Fries (1832). In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 823–858. [Google Scholar] [CrossRef]
- Kurtzman, C.P. Protomyces Unger (1833). In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 725–731. [Google Scholar] [CrossRef]
- Goto, S.; Sugiyama, J.; Hamamoto, M.; Komagata, K. Saitoella, a new anamorph genus in the Cryptococcaceae to accommodate two himalayan yeast isolates formerly identified as Rhodotorula glutinis. J. Gen. Appl. Microbiol. 1987, 33, 75–85. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Saitoella coloradoensis sp. nov., a new species of the Ascomycota, subphylum Taphrinomycotina. Antonie van Leeuwenhoek 2012, 101, 795–802. [Google Scholar] [CrossRef][Green Version]
- Sjamsuridza, W.; Tajiri, Y.; Nishida, H.; Thuan, T.B.; Kawasaki, H.; Hirata, A.; Yokota, A.; Sugiyama, J. Evolutionary relationships of members of the genera Taphrina, Protomyces, Schizosaccharomyces, and related taxa within the Archiascomycetes: Integrated analysis of genotypic and phenotypic characters. Mycoscience 1997, 38, 267–280. [Google Scholar] [CrossRef]
- Kurtzman, C.P. Discussion of teleomorphic and anamorphic ascomycetous yeasts and yeast-like taxa. In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 293–307. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Sugiyama, J. Saccharomycotina and Taphrinomycotina: The yeasts and yeastlike fungi of the Ascomycota. In The Mycota (A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research), Systematics and Evolution, 2nd ed.; McLaughlin, D., Spatafora, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 7B, pp. 3–33. [Google Scholar] [CrossRef]
- Reddy, M.S.; Kramer, C.L. A taxonomic revision of the Protomycetales. Mycotaxon 1975, 3, 1–50. [Google Scholar]
- The National Center for Biotechnology Information. Nucleotide. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 10 December 2020).
- Nguyen, T.A.; Cissé, O.H.; Wong, J.Y.; Zheng, P.; Hewitt, D.; Nowrousian, M.; Stajich, J.E.; Jedd, G. Innovation and constraint leading to complex multicellularity in the Ascomycota. Nat. Commun. 2017, 8, 14444. [Google Scholar] [CrossRef]
- Nagy, L.G. Evolution: Complex Multicellular Life with 5500 Genes. Curr. Biol. 2017, 27, R609–R612. [Google Scholar] [CrossRef]
- Rosling, A.; Cox, F.; Cruz-Martinez, K.; Ihrmark, K.; Grelet, G.-A.; Lindahl, B.D.; Menkis, A.; James, T.Y. Archaeorhizomycetes: Unearthing an Ancient Class of Ubiquitous Soil Fungi. Science 2011, 333, 876–879. [Google Scholar] [CrossRef]
- Menkis, A.; Urbina, H.; James, T.Y.; Rosling, A. Archaeorhizomyces borealis sp. nov. and a sequence-based classification of related soil fungal species. Fungal Biol. 2014, 118, 943–955. [Google Scholar] [CrossRef]
- Schadt, C.W.; Martin, A.P.; Lipson, D.A.; Schmidt, S.K. Seasonal Dynamics of Previously Unknown Fungal Lineages in Tundra Soils. Science 2003, 301, 1359–1361. [Google Scholar] [CrossRef]
- Porter, T.M.; Schadt, C.W.; Rizvi, L.; Martin, A.P.; Schmidt, S.K.; Scott-Denton, L.; Vilgalys, R.; Moncalvo, J.M. Widespread occurrence and phylogenetic placement of a soil clone group adds a prominent new branch to the fungal tree of life. Mol. Phylogenet. Evol. 2008, 46, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Zullo, B.A.; Ciafardini, G. Virgin Olive Oil Quality Is Affected by the Microbiota that Comprise the Biotic Fraction of the Oil. Microorganisms 2020, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Buckland, G.; González, C.A. Trends in olive oil production, supply and consumption in Mediterranean countries from 1961 to the present day. In Olives and Olive Oil in Health and Disease Prevention; Preedy, V.R., Watson, R.R., Eds.; Academic Press: London, UK, 2010; pp. 689–698. [Google Scholar] [CrossRef]
- Preedy, V.R.; Watson, R.R. (Eds.) Olives and Olive Oil in Health and Disease Prevention; Academic Press: London, UK, 2010. [Google Scholar]
- Ciafardini, G.; Zullo, B.A. Microbiological activity in stored olive oil. Int. J. Food Microbiol. 2002, 75, 111–118. [Google Scholar] [CrossRef]
- Čadež, N.; Raspor, P.; Turchetti, B.; Cardinali, G.; Ciafardini, G.; Veneziani, G.; Péter, G. Candida adriatica sp. nov. and Candida molendinolei sp. nov., two yeast species isolated from olive oil and its by-products. Int. J. Syst. Evol. Microbiol. 2012, 62, 2296–2302. [Google Scholar] [CrossRef] [PubMed]
- Čadež, N.; Dlauchy, D.; Raspor, P.; Péter, G. Ogataea kolombanensis sp. nov., Ogataea histrianica sp. nov. and Ogataea deakii sp. nov., three novel yeast species from plant sources. Int. J. Syst. Evol. Microbiol. 2013, 63, 3115–3123. [Google Scholar] [CrossRef]
- Péter, G.; Dlauchy, D.; Tóbiás, A.; Fülöp, L.; Podgoršek, M.; Čadež, N. Brettanomyces acidodurans sp. nov., a new acetic acid producing yeast species from olive oil. Antonie van Leeuwenhoek 2017, 110, 657–664. [Google Scholar] [CrossRef]
- Čadež, N.; Dlauchy, D.; Tóbiás, A.; Péter, G. Kuraishia mediterranea sp. nov., a methanol-assimilating yeast species from olive oil and its sediment. Int. J. Syst. Evol. Microbiol. 2017, 67, 4846–4850. [Google Scholar] [CrossRef]
- Ciafardini, G.; Zullo, B.A.; Antonielli, L.; Corte, L.; Roscini, L.; Cardinali, G. Yamadazyma terventina sp. nov., a yeast species of the Yamadazyma clade from Italian olive oils. Int. J. Syst. Evol. Microbiol. 2013, 63, 372–376. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Fell, J.W.; Boekhout, T.; Robert, V. Methods for isolation, phenotypic characterization and maintenance of yeasts. In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 1, pp. 87–110. [Google Scholar] [CrossRef]
- Nirenberg, H.I. Untersuchungen über die morphologische und biologische Differenzierung in der Fusarium Sektion Liseola. Mitt. Biol. Bund. Land. Forst. 1976, 169, 1–117. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie van Leeuwenhoek 1998, 73, 331–371. [Google Scholar] [CrossRef]
- Dlauchy, D.; Tornai-Lehoczki, J.; Sedláček, I.; Audy, M.; Péter, G. Debaryomyces psychrosporus sp. nov., a yeast species from a Venezuelan cave. Antonie van Leeuwenhoek 2010, 99, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Péter, G.; Tornai-Lehoczki, J.; Dlauchy, D. Candida ogatae sp. nov., an anamorphic member of the Kuraishia clade. FEMS Yeast Res. 2009, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Schwartz, K.; Sherlock, G. Preparation of Yeast DNA Sequencing Libraries. Cold Spring Harb. Protoc. 2016, 10, 871–876. [Google Scholar] [CrossRef]
- Yue, J.-X.; Liti, G. Long-read sequencing data analysis for yeasts. Nat. Protoc. 2018, 13, 1213–1231. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Weiß, C.L.; Pais, M.; Cano, L.M.; Kamoun, S.; Burbano, H.A. nQuire: A statistical framework for ploidy estimation using next generation sequencing. BMC Bioinform. 2018, 19, 1–8. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- The National Center for Biotechnology Information. Genomes. Available online: https://www.ncbi.nlm.nih.gov/home/genomes (accessed on 27 October 2020).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Salichos, L.; Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nat. Cell Biol. 2013, 497, 327–331. [Google Scholar] [CrossRef]
- Salichos, L.; Stamatakis, A.; Rokas, A. Novel Information Theory-Based Measures for Quantifying Incongruence among Phylogenetic Trees. Mol. Biol. Evol. 2014, 31, 1261–1271. [Google Scholar] [CrossRef]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 15–30. [Google Scholar] [CrossRef]
- Lock, A.; Rutherford, K.; Harris, M.A.; Hayles, J.; Oliver, S.G.; Bähler, J.; Wood, V. PomBase 2018: User-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2019, 47, D821–D827. [Google Scholar] [CrossRef]
- RefSeq: NCBI Reference Sequence Database. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 30 October 2019).
- Steenwyk, J.L.; Opulente, D.A.; Kominek, J.; Shen, X.-X.; Zhou, X.; Labella, A.; Bradley, N.P.; Eichman, B.F.; Čadež, N.; Libkind, D.; et al. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 2019, 17, e3000255. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Robnett, C.J. Relationships among genera of the Saccharomycotina (Ascomycota) from multigene phylogenetic analysis of type species. FEMS Yeast Res. 2013, 13, 23–33. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.H.; Huang, D.W.; Kutty, G.; Ishihara, M.; Wang, H.; Abouelleil, A.; Bishop, L.; Davey, E.; Deng, R.; et al. Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat. Commun. 2016, 7, 10740. [Google Scholar] [CrossRef] [PubMed]
- Jeffares, D.C.; Rallis, C.; Rieux, A.; Speed, D.; Převorovský, M.; Mourier, T.; Marsellach, X.; Iqbal, Z.; Lau, W.; Cheng, T.M.; et al. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat. Genet. 2015, 47, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Aliouat, E.M.; Standaert-Vitse, A.; Werkmeister, E.; Pottier, M.; Pinçon, C.; Dei-Cas, E.; Aliouat-Denis, C.-M. Ploidy of cell-sorted trophic and cystic forms of Pneumocystis carinii. PLoS ONE 2011, 6, e20935. [Google Scholar] [CrossRef] [PubMed]
- Albalat, R.; Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef]
- Shen, X.-X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Watanabe, H.; Lipman, D.J.; Koonin, E.V. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc. Natl. Acad. Sci. USA 2000, 97, 11319–11324. [Google Scholar] [CrossRef] [PubMed]
- Katinka, M.D.; Duprat, S.; Cornillot, E.; Méténier, G.; Thomarat, F.; Prensier, G.; Barbe, V.; Peyretaillade, E.; Brottier, P.; Wincker, P.; et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nat. Cell Biol. 2001, 414, 450–453. [Google Scholar] [CrossRef]
- Almeida, J.M.G.C.F.; Cissé, O.H.; Fonseca, A.; Pagni, M.; Hauser, P.M. Comparative Genomics Suggests Primary Homothallism of Pneumocystis Species. mBio 2015, 6, 02250–02214. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Fell, J.W.; Boekhout, T. (Eds.) The Yeasts: A Taxonomic Study, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2011; Volumes 1–3, p. 2080. [Google Scholar]
- Sugiyama, J.; Hamamoto, M. Goto, Sugiyama, Hamamoto & Komagata (1987). In The Yeasts: A Taxonomic Study, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 2, pp. 1313–1315. [Google Scholar] [CrossRef]
- Haase, M.A.B.; Kominek, J.; Langdon, Q.K.; Kurtzman, C.P.; Hittinger, C.T. Genome sequence and physiological analysis of Yamadazyma laniorum f.a. sp. nov. and a reevaluation of the apocryphal xylose fermentation of its sister species, Candida tenuis. FEMS Yeast Res. 2017, 17, 1–13. [Google Scholar] [CrossRef]
- Čadež, N.; Bellora, N.; Ulloa, R.; Hittinger, C.T.; Libkind, D. Genomic content of a novel yeast species Hanseniaspora gamundiae sp. nov. from fungal stromata (Cyttaria) associated with a unique fermented beverage in Andean Patagonia, Argentina. PLoS ONE 2019, 14, e0210792. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Fujita, Y.; Tanaka, N.; Tohda, H.; Itadani, A.; Takegawa, K. New insights into galactose metabolism by Schizosaccharomyces pombe: Isolation and characterization of a galactose-assimilating mutant. J. Biosci. Bioeng. 2011, 111, 158–166. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Name | Strain/Specimen * | GenBank Accession Numbers |
|---|---|---|
| Archaeorhizomyces borealis | NS99-600T | KF993708 |
| Archaeorhizomyces finlayi | CBS 128710T | NG_064244 |
| Emericella nidulans | ATCC 10074T | NG_055739 |
| Filobasidiella neoformans | CBS 132T | KY107233 |
| Lipomyces starkeyi | NRRL Y-11557T | DQ518981 |
| Neolecta irregularis | ZW-Geo79-Clark | AY789380 |
| Neolecta vitellina | DJM1533V | KC333874 |
| Neournula pouchetii | MO-205345V | KT968655 |
| Neurospora crassa | NRRL 13141 | U40124 |
| Novakomyces olei | NCAIM Y.02187T | MG250349 |
| Ogataea parapolymorpha | NRRL YB-1982T | EU011621 |
| Pneumocystis carinii | / | M86760 |
| Pneumocystis jirovecii | RU7 | XR_001936353 |
| Pneumocystis murina | B123 | XR_486831 |
| Protomyces lactucaedebilis | NRRL YB-4353A | U84343 |
| Protomyces macrosporus | NRRL Y-12879A | U94939 |
| Rhodosporidium toruloides | CBS 6016T | KY109167 |
| Saccharomyces cerevisiae | NRRL Y-12632T | AY048154 |
| Saitoella coloradoensis | NRRL YB-2330T | JN161157 |
| Saitoella complicata | NRRL Y-17804T | JQ689076 |
| Schizosaccharomyces cryophilus | OY26T | GU470882 |
| Schizosaccharomyces japonicus | NRRL Y-1361T | U94943 |
| Schizosaccharomyces octosporus | NRRL Y-855T | U76525 |
| Schizosaccharomyces osmophilus | SZ134-FG-AT | MK253005 |
| Schizosaccharomyces pombe | NRRL Y-12796T | AY048171 |
| Taphrina deformans | CBS 356.35T | MH867217 |
| Taphrina populina | CBS 337.55T | AF492050 |
| Tuber melanosporum | GB200V | JQ925703 |
| Ustilago maydis | CBS 504.76A | L20287 |
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (Mb) | 14.3 | |
| GC content (%) | 43.8% | |
| Assembly statistics | ||
| Number of scaffolds | 15 | |
| N50 (kb) | 1688 | |
| L50 | 4 | |
| Number of unknown bases (N) | 0 | |
| Annotation statistics | ||
| Number of protein-coding genes | 3779 | |
| Gene length (median, bp) | 1694 | 22.2% of genome size |
| Exons per gene (mean) | 5.7 | − |
| Exon length (median, bp) | 1047 | 12.5% of genome size |
| Intron length (median, bp) | 261 | 5.7% of genome size |
| rDNA loci | 11 | − |
| tRNA genes | 75 | − |
| Repeats (bp) | 741,619 | 5.2% of genome size |
| Class I: Retrotransposons 1 | ||
| LTR transposons | 622 | 7.7% of repeats |
| Non-LTR transposons (LINE, SINE) | 177 | 2.2% of repeats |
| Class II: DNA transposons | 269 | 3.3% of repeats |
| Simple repeats | 5913 | 73.8% of repeats |
| A and GA-rich regions | 976 | 12.1% of repeats |
| Other | 63 | 0.8% of repeats |
| Complete BUSCO orthologs2 | 1063 | 80.9% out of 1315 |
| Duplicated BUSCOs | 2 | 0.2% |
| Fragmented BUSCOs | 75 | 5.7% |
| Missing BUSCOs | 177 | 13.4% |
| Genus | Urease | DBB | Starch Formation | CoQ | Carotenoid Pigments | Nitrate Assimilation | Fermentation | Sexual Reproduction in Pure Culture |
|---|---|---|---|---|---|---|---|---|
| Novakomyces | + | − | + | 10 | − | − | − | + |
| Taphrina | + | − | + | 10 | + | + | − | − |
| Protomyces | v | − | v | 10 | + | + (−) | − | − |
| Saitoella | + | − | − | 10 | + | + | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čadež, N.; Dlauchy, D.; Tome, M.; Péter, G. Novakomyces olei sp. nov., the First Member of a Novel Taphrinomycotina Lineage. Microorganisms 2021, 9, 301. https://doi.org/10.3390/microorganisms9020301
Čadež N, Dlauchy D, Tome M, Péter G. Novakomyces olei sp. nov., the First Member of a Novel Taphrinomycotina Lineage. Microorganisms. 2021; 9(2):301. https://doi.org/10.3390/microorganisms9020301
Chicago/Turabian StyleČadež, Neža, Dénes Dlauchy, Miha Tome, and Gábor Péter. 2021. "Novakomyces olei sp. nov., the First Member of a Novel Taphrinomycotina Lineage" Microorganisms 9, no. 2: 301. https://doi.org/10.3390/microorganisms9020301
APA StyleČadež, N., Dlauchy, D., Tome, M., & Péter, G. (2021). Novakomyces olei sp. nov., the First Member of a Novel Taphrinomycotina Lineage. Microorganisms, 9(2), 301. https://doi.org/10.3390/microorganisms9020301