Investigating Both Mucosal Immunity and Microbiota in Response to Gut Enteritis in Yellowtail Kingfish

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. Library Preparation and Sequencing
2.3. Bioinformatics and Statistical Analysis
3. Results
3.1. Analysis of the Gut and Skin Microbiota
3.1.1. Influence of Gut Enteritis on the Global Gastrointestinal and Skin Mucosal Microbiota
3.1.2. Taxonomic Composition and Potential Biomarkers of Gut Enteritis in the Gut and Skin Microbiota
3.2. Analysis of the Transcriptomics Data
3.2.1. Differential Expression in the Gut of Fish Exhibiting Different Health States
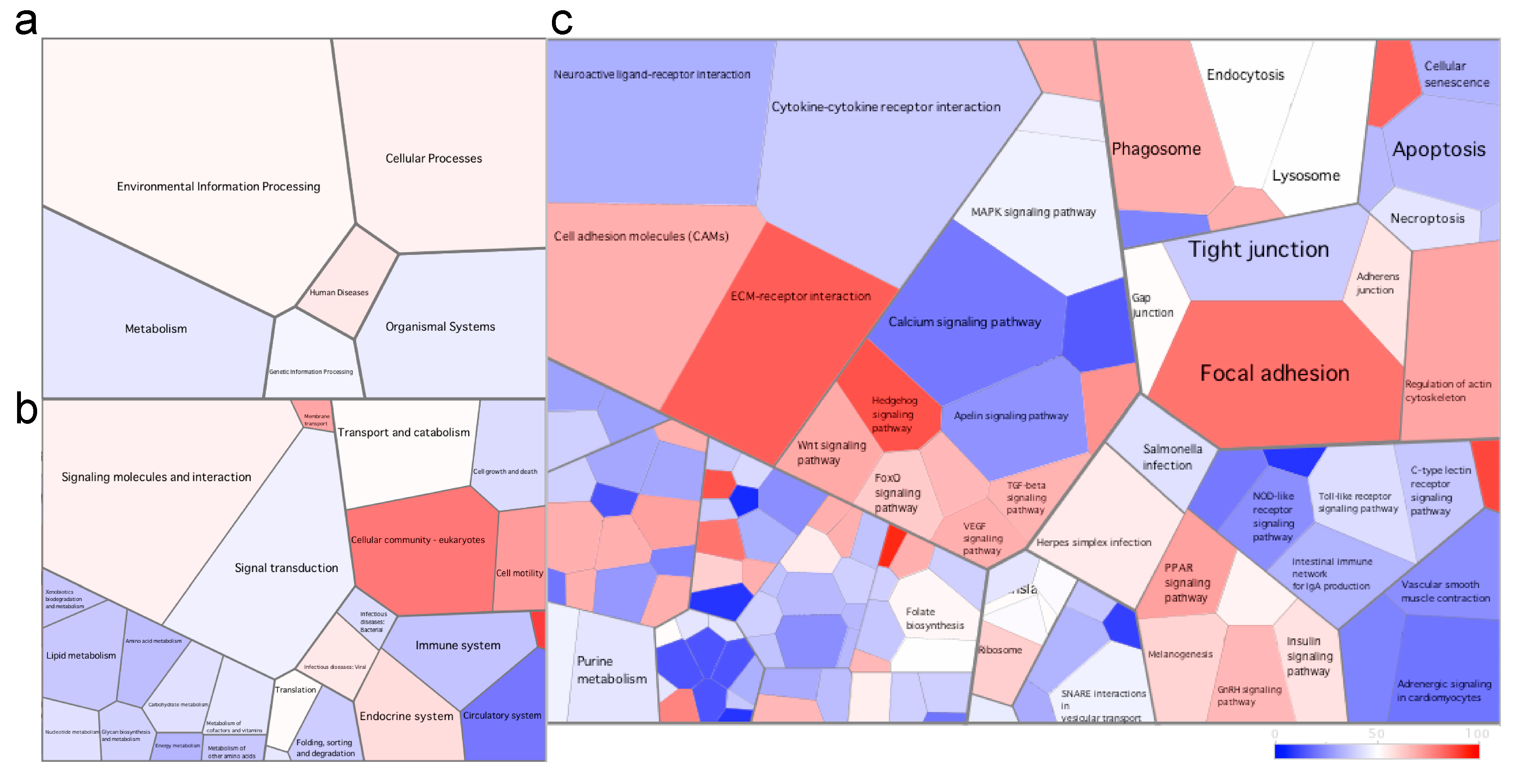
3.2.2. Differential Gene Expression and Associated Pathways in the Skin of Fish at the Late Stage of the Disease
3.2.3. Differential Gene Expression and Associated Pathways in the Skin of Fish at the early Stage of the Disease
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cabillon, N.A.R.; Lazado, C.C. Mucosal barrier functions of fish under changing environmental conditions. Fishes 2019, 4, 2. [Google Scholar] [CrossRef]
- Kelly, C.; Salinas, I. Under pressure: Interactions between commensal microbiota and the teleost immune system. Front. Immunol. 2017, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Arques, A.; De Schryver, P.; Bossier, P.; Gorkiewicz, G.; Mulero, V.; Gatlin, D.M.; Galindo-Villegas, J. Selective manipulation of the gut microbiota improves immune status in vertebrates. Front. Immunol. 2015, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.L.; Volkoff, H. Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 2019, 10, 12. [Google Scholar] [CrossRef]
- Gomez, D.; Sunyer, J.O.; Salinas, I. The mucosal immune system of fish: The evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 2013, 35, 1729–1739. [Google Scholar] [CrossRef]
- Salinas, I.; Parra, D. Fish mucosal immunity: Intestine. Mucosal Health Aquac. 2015, 135–158. [Google Scholar] [CrossRef]
- Booman, M.; Forster, I.; Vederas, J.C.; Groman, D.B.; Jones, S.R.M. Soybean meal-induced enteritis in atlantic salmon (Salmo salar) and chinook salmon (Oncorhynchus tshawytscha) but not in pink salmon (O. gorbuscha). Aquaculture 2018, 483, 238–243. [Google Scholar] [CrossRef]
- Bansemer, M.S.; Forder, R.E.A.; Howarth, G.S.; Suitor, G.M.; Bowyer, J.; Stone, D.A.J. The effect of dietary soybean meal and soy protein concentrate on the intestinal mucus layer and development of subacute enteritis in Yellowtail Kingfish (Seriola lalandi) at suboptimal water temperature. Aquac. Nutr. 2015, 21, 300–310. [Google Scholar] [CrossRef]
- Gu, M.; Bai, N.; Zhang, Y.Q.; Krogdahl, A. Soybean meal induces enteritis in turbot Scophthalmus maximus at high supplementation levels. Aquaculture 2016, 464, 286–295. [Google Scholar] [CrossRef]
- Uran, P.A.; Goncalves, A.A.; Taverne-Thiele, J.J.; Schrama, J.W.; Verreth, J.A.J.; Rombout, J. Soybean meal induces intestinal inflammation in common carp (Cyprinus carpio L.). Fish Shellfish Immunol. 2008, 25, 751–760. [Google Scholar] [CrossRef]
- Coronado, M.; Solis, C.J.; Hernandez, P.P.; Feijoo, C.G. Soybean meal-induced intestinal inflammation in zebrafish is t cell-dependent and has a th17 cytokine profile. Front. Immunol. 2019, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tan, B.P.; Ye, G.L.; Wang, J.X.; Dong, X.H.; Yang, Q.H.; Chi, S.Y.; Liu, H.Y.; Zhang, S.; Zhang, H.T. Identification of potential biomarkers for soybean meal-induced enteritis in juvenile pearl gentian grouper, Epinephelus lanceolatus male x Epinephelus fuscoguttatus female. Aquaculture 2019, 512, 14. [Google Scholar] [CrossRef]
- Viana, M.T.; Rombenso, A.N.; Del Rio-Zaragoza, O.B.; Nomura, M.; Diaz-Arguello, R.; Mata-Sotres, J.A. Intestinal impairment of the California yellowtail, Seriola dorsalis, using soybean meal in the diet. Aquaculture 2019, 513, 8. [Google Scholar] [CrossRef]
- Krol, E.; Douglas, A.; Tocher, D.R.; Crampton, V.O.; Speakman, J.R.; Secombes, C.J.; Martin, S.A.M. Differential responses of the gut transcriptome to plant protein diets in farmed Atlantic salmon. BMC Genom. 2016, 17, 16. [Google Scholar] [CrossRef]
- Martin, S.A.M.; Krol, E. Nutrigenomics and immune function in fish: New insights from omics technologies. Dev. Comp. Immunol. 2017, 75, 86–98. [Google Scholar] [CrossRef]
- Kong, W.G.; Huang, C.; Tang, Y.; Zhang, D.; Wu, Z.X.; Chen, X.X. Effect of Bacillus subtilis on Aeromonas hydrophila-induced intestinal mucosal barrier function damage and inflammation in grass carp (Ctenopharyngodon idella). Sci. Rep. 2017, 7, 11. [Google Scholar] [CrossRef]
- Sitja-Bobadilla, A.; Gil-Solsona, R.; Estensoro, I.; Piazzon, M.C.; Martos-Sitcha, J.A.; Picard-Sanchez, A.; Fuentes, J.; Sancho, J.V.; Calduch-Giner, J.A.; Hernandez, F.; et al. Disruption of gut integrity and permeability contributes to enteritis in a fish-parasite model: A story told from serum metabolomics. Parasites Vectors 2019, 12, 18. [Google Scholar] [CrossRef]
- Gaulke, C.A.; Martins, M.L.; Watral, V.G.; Humphreys, I.R.; Spagnoli, S.T.; Kent, M.L.; Sharpton, T.J. A longitudinal assessment of host-microbe-parasite interactions resolves the zebrafish gut microbiome’s link to Pseudocapillaria tomentosa infection and pathology. Microbiome 2019, 7, 16. [Google Scholar] [CrossRef]
- Brugman, S. The zebrafish as a model to study intestinal inflammation. Dev. Comp. Immunol. 2016, 64, 82–92. [Google Scholar] [CrossRef]
- Tran, N.T.; Zhang, J.; Xiong, F.; Wang, G.T.; Li, W.X.; Wu, S.G. Altered gut microbiota associated with intestinal disease in grass carp (Ctenopharyngodon idellus). World J. Microbiol. Biotechnol. 2018, 34, 9. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Catalano, S.R.; Wos-Oxley, M.L.; Stephens, F.; Landos, M.; Bansemer, M.S.; Stone, D.A.J.; Qin, J.G.; Oxley, A.P.A. The Inner Workings of the Outer Surface: Skin and Gill Microbiota as Indicators of Changing Gut Health in Yellowtail Kingfish. Front. Microbiol. 2018, 8, 17. [Google Scholar] [CrossRef]
- Zheng, D.P.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.J.; Huffnagle, G.B. The microbiome and regulation of mucosal immunity. Immunology 2014, 142, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Lescak, E.A.; Milligan-Myhre, K.C. Teleosts as model organisms to understand host-microbe interactions. J. Bacteriol. 2017, 199, 11. [Google Scholar] [CrossRef]
- Marjara, I.S.; Chikwati, E.M.; Valen, E.C.; Krogdahl, A.; Bakke, A.M. Transcriptional regulation of IL-17A and other inflammatory markers during the development of soybean meal-induced enteropathy in the distal intestine of Atlantic salmon (Salmo salar L.). Cytokine 2012, 60, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. A microbial sea of possibilities: Current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2019, 12, 1101–1134. [Google Scholar] [CrossRef]
- Zhang, J.J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, 11. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vazquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, 17. [Google Scholar] [CrossRef]
- Santamaria, R.; Pierre, P. Voronto: Mapper for expression data to ontologies. Bioinformatics 2012, 28, 2281–2282. [Google Scholar] [CrossRef][Green Version]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Gu, M.; Bai, N.; Xu, B.Y.; Xu, X.J.; Jia, Q.; Zhang, Z.Y. Protective effect of glutamine and arginine against soybean meal-induced enteritis in the juvenile turbot (Scophthalmus maximus). Fish Shellfish Immunol. 2017, 70, 95–105. [Google Scholar] [CrossRef]
- Tan, C.; Zhou, H.; Wang, X.; Mai, K.; He, G. Resveratrol attenuates oxidative stress and inflammatory response in turbot fed with soybean meal based diet. Fish Shellfish Immunol. 2019, 91, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, P.E.; Solis, C.J.; De la Paz, J.F.; Alaurent, T.G.S.; Caruffo, M.; Hernandez, A.J.; Dantagnan, P.; Feijoo, C.G. Lactoferrin decreases the intestinal inflammation triggered by a soybean meal-based diet in zebrafish. J. Immunol. Res. 2016, 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Tello, K.; Ehrenfeld, N.; Solis, C.J.; Ulloa, P.E.; Hedrera, M.; Pizarro-Guajardo, M.; Paredes-Sabja, D.; Feijoo, C.G. Effect of microalgae on intestinal inflammation triggered by soybean meal and bacterial infection in zebrafish. PLoS ONE 2017, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Romarheim, O.H.; Overland, M.; Mydland, L.T.; Skrede, A.; Landsverk, T. Bacteria grown on natural gas prevent soybean meal-induced enteritis in Atlantic salmon. J. Nutr. 2011, 141, 124–130. [Google Scholar] [CrossRef]
- Li, C.; Zhang, B.; Wang, X.; Pi, X.; Wang, X.; Zhou, H.; Mai, K.; He, G. Improved utilization of soybean meal through fermentation with commensal Shewanella sp. MR-7 in turbot (Scophthalmus maximus L.). Microbial Cell Factories 2019, 18, 214. [Google Scholar] [CrossRef]
- Abernathy, J.; Brezas, A.; Snekvik, K.R.; Hardy, R.W.; Overturf, K. Integrative functional analyses using rainbow trout selected for tolerance to plant diets reveal nutrigenomic signatures for soy utilization without the concurrence of enteritis. PLoS ONE 2017, 12, 30. [Google Scholar] [CrossRef]
- Dam, C.T.M.; Elizur, A.; Ventura, T.; Salini, M.; Smullen, R.; Pirozzi, I.; Booth, M. Apparent digestibility of raw materials by yellowtail kingfish (Seriola lalandi). Aquaculture 2019, 511, 11. [Google Scholar] [CrossRef]
- Lowrey, L.; Woodhams, D.C.; Tacchi, L.; Salinas, I. Topographical mapping of the rainbow trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community with antifungal properties in the skin. Appl. Environ. Microbiol. 2015, 81, 6915–6925. [Google Scholar] [CrossRef]
- Fu, P.P.; Xiong, F.; Feng, W.W.; Zou, H.; Wu, S.G.; Li, M.; Wang, G.T.; Li, W.X. Effect of intestinal tapeworms on the gut microbiota of the common carp, Cyprinus carpio. Parasites Vectors 2019, 12, 11. [Google Scholar] [CrossRef]
- Jin, Y.; Angell, I.L.; Rod Sandve, S.; Snipen, L.G.; Olsen, Y.; Rudi, K. Atlantic salmon raised with diets low in long-chain polyunsaturated n-3 fatty acids in freshwater have a Mycoplasma-dominated gut microbiota at sea. Aquac. Environ. Interact. 2019, 11, 31–39. [Google Scholar] [CrossRef]
- Nishikawa, M.S.; Nakane, D.; Toyonaga, T.; Kawamoto, A.; Kato, T.; Namba, K.; Miyata, M. Refined mechanism of Mycoplasma mobile gliding based on structure, ATPase activity, and sialic acid binding of machinery. mBio 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.-A.; Yan, G.-Y.; Huang, J.-M.; Danchin, A.; Wang, Y.; He, L.-S. Genomic characterization of a novel gut symbiont from the Hadal snailfish. Front. Microbiol. 2020, 10, 2978. [Google Scholar] [CrossRef] [PubMed]
- Sylvain, F.-É.; Holland, A.; Bouslama, S.; Audet-Gilbert, É.; Lavoie, C.; Val, A.L.; Derome, N. Fish skin and gut microbiomes show contrasting signatures of host species and habitat. Appl. Environ. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- De Schryver, P.; Vadstein, O. Ecological theory as a foundation to control pathogenic invasion in aquaculture. Isme J. 2014, 8, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, N.; Coll, J.; Salguero, F.J.; Tafalla, C. Co-injection of interleukin 8 with the glycoprotein gene from viral haemorrhagic septicemia virus (VHSV) modulates the cytokine response in rainbow trout (Oncorhynchus mykiss). Vaccine 2006, 24, 5615–5626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.T.; Ding, L.G.; Yu, Y.Y.; Kong, W.G.; Yin, Y.X.; Huang, Z.Y.; Zhang, X.Z.; Xu, Z. The change of teleost skin commensal microbiota is associated with skin mucosal transcriptomic responses during parasitic infection by ichthyophthirius multifillis. Front. Immunol. 2018, 9, 16. [Google Scholar] [CrossRef]
- Stadtmann, A.; Zarbock, A. CXCR2: From bench to bedside. Front. Immunol. 2012, 3, 12. [Google Scholar] [CrossRef]
- Lee, J.-W.; Yang, H.; Noh, J.K.; Kim, H.C.; Park, C.-J.; Park, J.-W.; Hwang, I.J.; Kim, S.Y.; Lee, J.-H. RAG-1 and IgM genes, markers for early development of the immune system in olive flounder, Paralichthys olivaceus. Dev. Reprod. 2014, 18, 99. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Ángeles Esteban, M. An overview of the immunological defenses in fish skin. ISRN Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Lo, J.H.; Lin, C.M.; Chen, M.J.; Chen, T.T. Altered gene expression patterns of innate and adaptive immunity pathways in transgenic rainbow trout harboring Cecropin P1 transgene. BMC Genom. 2014, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Caltagarone, J.; Jing, Z.; Bowser, R. Focal adhesions regulate Aβ signaling and cell death in Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2007, 1772, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-L.; Chen, L.-C.; Shen, T.-L. Emerging roles of focal adhesion kinase in cancer. Biomed. Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; Ginsberg, M.H. Networks and crosstalk: Integrin signalling spreads. Nat. Cell Biol. 2002, 4, E65–E68. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-D.; Jiang, W.-D.; Wu, P.; Liu, Y.; Jiang, J.; Tan, B.-P.; Yang, Q.-H.; Kuang, S.-Y.; Tang, L.; Zhou, X.-Q. Soybean β-conglycinin caused intestinal inflammation and oxidative damage in association with NF-κB, TOR and Nrf2 in juvenile grass carp (Ctenopharyngodon idella): Varying among different intestinal segments. Fish Shellfish Immunol. 2019, 95, 105–116. [Google Scholar] [CrossRef]
- Dong, S.; Ding, L.G.; Cao, J.F.; Liu, X.; Xu, H.Y.; Meng, K.F.; Yu, Y.Y.; Wang, Q.C.; Xu, Z. Viral-Infected change of the digestive tract microbiota associated with mucosal immunity in Teleost fish. Front. Immunol. 2019, 10, 13. [Google Scholar] [CrossRef]
- Limborg, M.T.; Alberdi, A.; Kodama, M.; Roggenbuck, M.; Kristiansen, K.; Gilbert, M.T.P. Applied hologenomics: Feasibility and potential in aquaculture. Trends Biotechnol. 2018, 36, 252–264. [Google Scholar] [CrossRef]
- Bjorstad, A.; Fu, H.M.; Karlsson, A.; Dahlgren, C.; Bylund, J. Interleukin-8-derived peptide has antibacterial activity. Antimicrob. Agents Chemother. 2005, 49, 3889–3895. [Google Scholar] [CrossRef]
- Zuyderduyn, S.; Ninaber, D.K.; Hiemstra, P.S.; Rabe, K.F. The antimicrobial peptide LL-37 enhances IL-8 release by human airway smooth muscle cells. J. Allergy Clin. Immunol. 2006, 117, 1328–1335. [Google Scholar] [CrossRef]
- Bridle, A.; Nosworthy, E.; Polinski, M.; Nowak, B. Evidence of an antimicrobial-immunomodulatory role of atlantic salmon cathelicidins during infection with Yersinia ruckeri. PLoS ONE 2011, 6, 11. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the human gut microbiome to inflammatory Cytokine production capacity. Cell 2016, 167, 1125–1136. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Gene Expression | HvsE | HvsL | EvsL |
|---|---|---|---|---|
| gut | upregulated | 15 | 31 | 18 |
| downregulated | 52 | 57 | 1 | |
| total | 67 | 88 | 19 | |
| skin | upregulated | 54 | 481 | 552 |
| downregulated | 130 | 986 | 1516 | |
| total | 184 | 1467 | 2068 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. Investigating Both Mucosal Immunity and Microbiota in Response to Gut Enteritis in Yellowtail Kingfish. Microorganisms 2020, 8, 1267. https://doi.org/10.3390/microorganisms8091267
Legrand TPRA, Wynne JW, Weyrich LS, Oxley APA. Investigating Both Mucosal Immunity and Microbiota in Response to Gut Enteritis in Yellowtail Kingfish. Microorganisms. 2020; 8(9):1267. https://doi.org/10.3390/microorganisms8091267
Chicago/Turabian StyleLegrand, Thibault P. R. A., James W. Wynne, Laura S. Weyrich, and Andrew P. A. Oxley. 2020. "Investigating Both Mucosal Immunity and Microbiota in Response to Gut Enteritis in Yellowtail Kingfish" Microorganisms 8, no. 9: 1267. https://doi.org/10.3390/microorganisms8091267
APA StyleLegrand, T. P. R. A., Wynne, J. W., Weyrich, L. S., & Oxley, A. P. A. (2020). Investigating Both Mucosal Immunity and Microbiota in Response to Gut Enteritis in Yellowtail Kingfish. Microorganisms, 8(9), 1267. https://doi.org/10.3390/microorganisms8091267