Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change

 , , ,
, , ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment of Subjects and Sampling
2.2. Data Collection
2.3. DNA Extraction, PCR Amplification, and Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
3.1. Characterstics of the Participants and Data Summary of Amplicon Sequencing
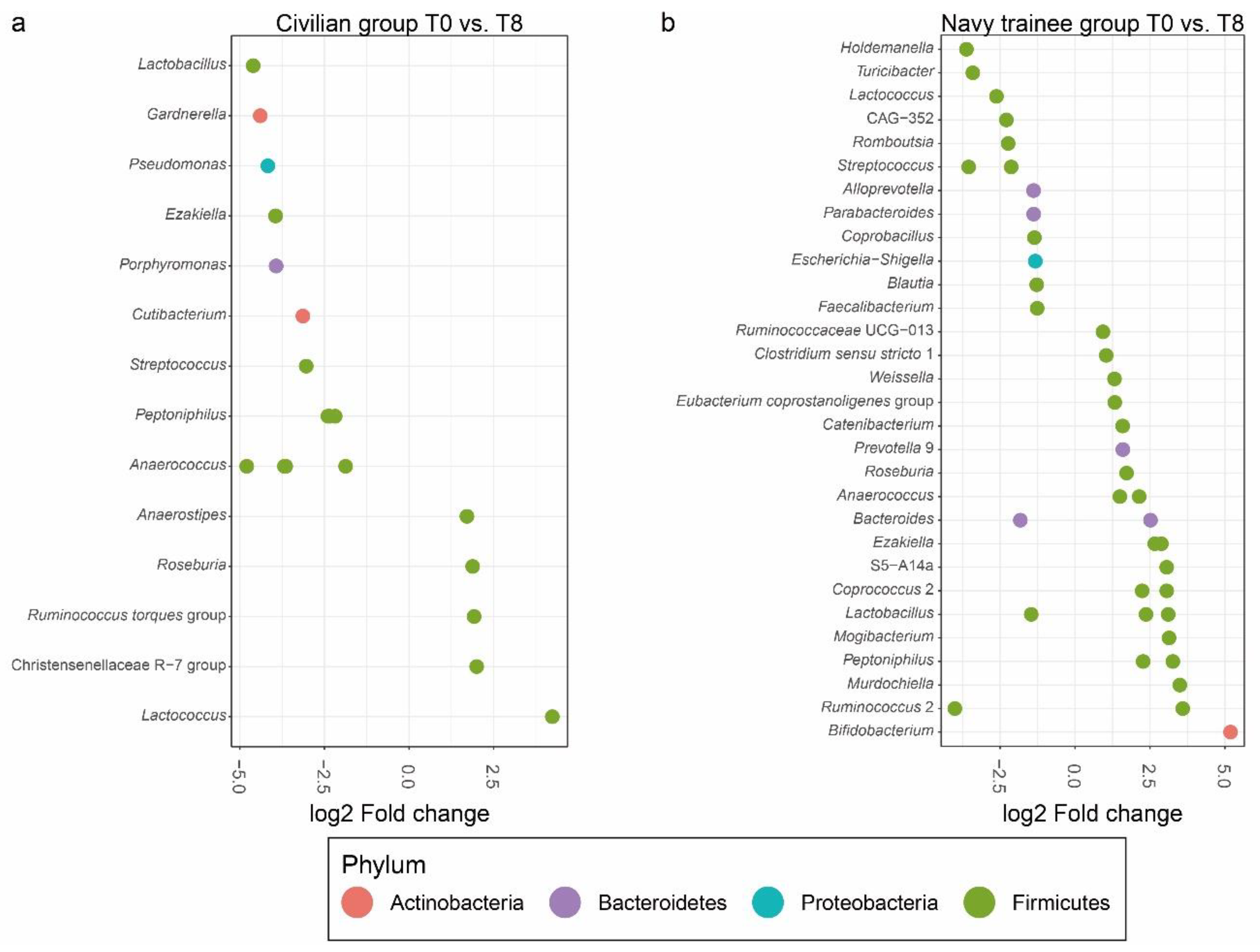
3.2. Intestinal Microbial Communities after Eight Weeks of Lifestyle Intervention
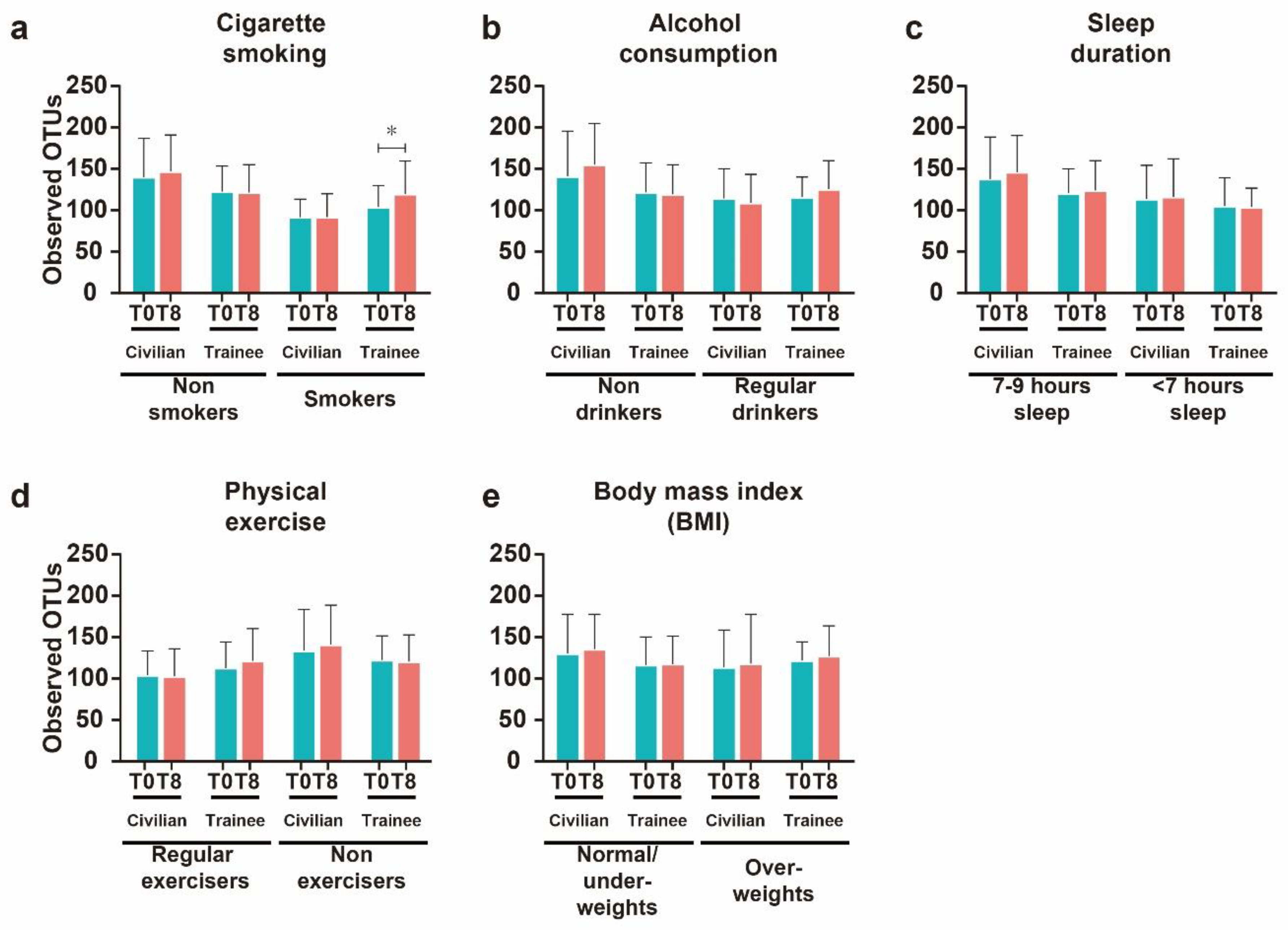
3.3. Effects of Modifiable Lifestyles Factors on Diversity of Intestinal Microbial Communities
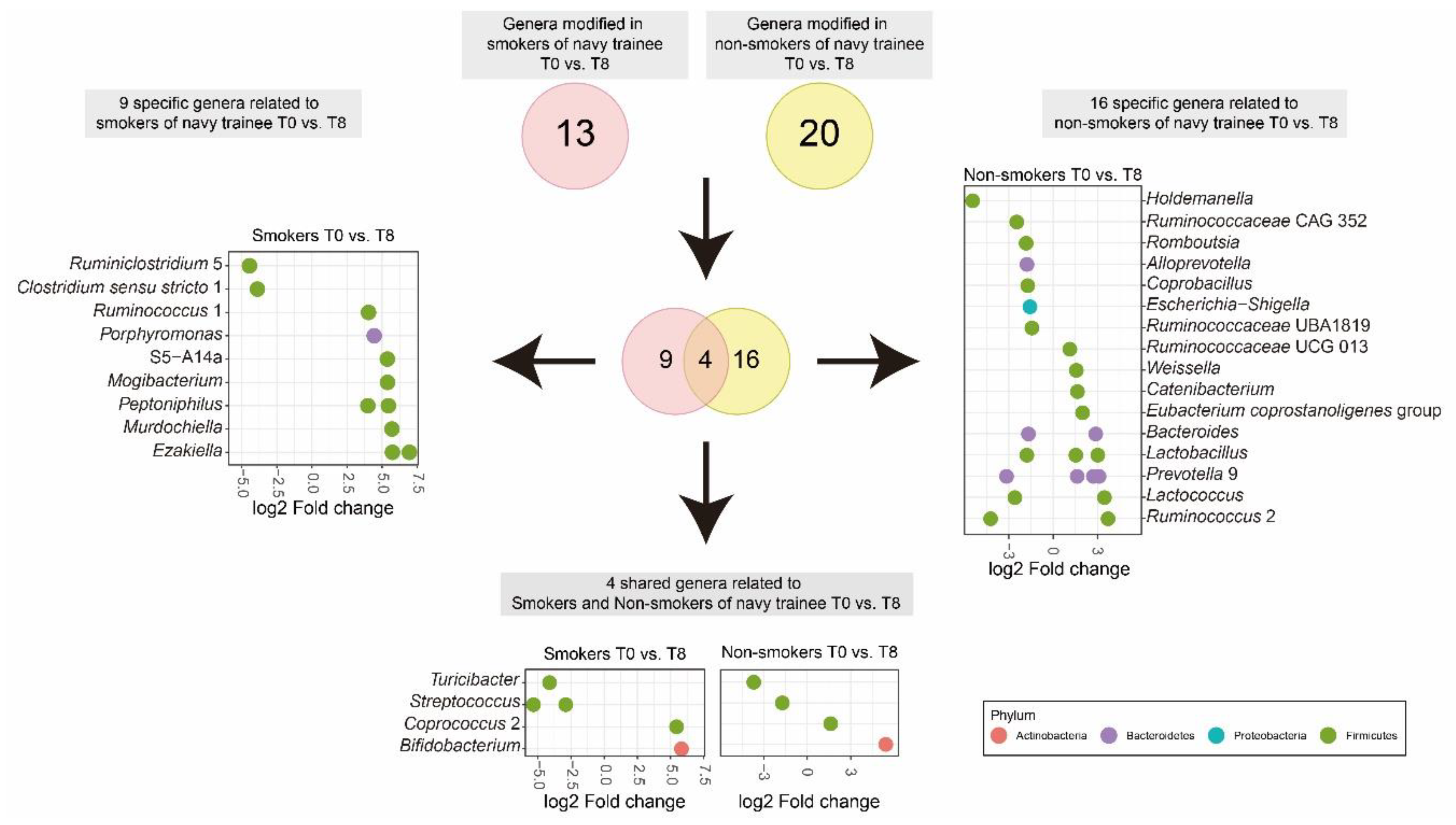
3.4. Cigarette Smoking Altered the Abundance of Individual Bacterial Taxa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Bengmark, S. Ecological control of the gastrointestinal tract. The role of probiotic flora. Gut 1998, 42, 2–7. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226. [Google Scholar] [CrossRef]
- Chow, J.; Lee, S.M.; Shen, Y.; Khosravi, A.; Mazmanian, S.K. Host-bacterial symbiosis in health and disease. Adv. Immunol. 2010, 107, 243–274. [Google Scholar]
- Karlsson, C.L.; Onnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The microbiota of the gut in preschool children with normal and excessive body weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Li, H.; Ni, C.; Du, Z.; Yan, F. Human oral microbiota and its modulation for oral health. Biomed. Pharmacother. 2018, 99, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 1999, 69, 1035S–1045S. [Google Scholar] [CrossRef]
- Mandar, R.; Mikelsaar, M. Transmission of mother’s microflora to the newborn at birth. Biol. Neonate 1996, 69, 30–35. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Zhong, H.; Penders, J.; Shi, Z.; Ren, H.; Cai, K.; Fang, C.; Ding, Q.; Thijs, C.; Blaak, E.E.; Stehouwer, C.D.A.; et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome 2019, 7, 2. [Google Scholar] [CrossRef]
- Mehta, R.S.; Abu-Ali, G.S.; Drew, D.A.; Lloyd-Price, J.; Subramanian, A.; Lochhead, P.; Joshi, A.D.; Ivey, K.L.; Khalili, H.; Brown, G.T.; et al. Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 2018, 3, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, T.N.; Hilton, C.; Tsai, P.; Apple, B.; Shepherd, P.; Cutfield, W.S.; O’Sullivan, J.M. Long-term stability in the gut microbiome over 46 years in the life of Billy Apple. Hum. Microbiome 2017, 5, 7–10. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Stress and the gut: Pathophysiology, clinical consequences, diagnostic approach and treatment options. J. Physiol. Pharmacol. 2011, 62, 591–599. [Google Scholar] [PubMed]
- Rauch, M.; Lynch, S. The potential for probiotic manipulation of the gastrointestinal microbiome. Curr. Opin. Biotechnol. 2012, 23, 192–201. [Google Scholar] [CrossRef]
- Lee, S.H.; Yun, Y.; Kim, S.J.; Lee, E.J.; Chang, Y.; Ryu, S.; Shin, H.; Kim, H.L.; Kim, H.N.; Lee, J.H. Association between cigarette smoking status and composition of gut microbiota: Population-based cross-sectional study. J. Clin. Med. 2018, 7, 282. [Google Scholar] [CrossRef]
- Ortiz-Alvarez, L.; Xu, H.; Martinez-Tellez, B. Influence of exercise on the human gut microbiota of healthy adults: A systematic review. Clin. Transl. Gastroen. 2020, 11, 2. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Starkel, P.; Windey, K.; Tremaroli, V.; Backhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Benedict, C.; Vogel, H.; Jonas, W.; Woting, A.; Blaut, M.; Schurmann, A.; Cedernaes, J. Gut microbiota and glucometabolic alterations in response to recurrent partial sleep deprivation in normal-weight young individuals. Mol. Metab. 2016, 5, 1175–1186. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Knight, R.; Leibel, R.L. The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol. Metab. 2015, 26, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Sutradhar, R.; Yao, Z.; Wodchis, W.P.; Rosella, L.C. Smoking, drinking, diet and physical activity—Modifiable lifestyle risk factors and their associations with age to first chronic disease. Int. J. Epidemiol. 2020, 49.1, 113–130. [Google Scholar] [CrossRef]
- Hirshkowitz, M.; Whiton, K.; Albert, S.M.; Alessi, C.; Bruni, O.; DonCarlos, L.; Hazen, N.; Herman, J.; Katz, E.S.; Kheirandish-Gozal, L.; et al. National sleep foundation’s sleep time duration recommendations: Methodology and results summary. Sleep Health 2015, 1, 40–43. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Xu, Z.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiom 2018, 6, 90. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.; Ludwig, W.; Peplies, J.; Glöckner, F. Silva: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- McMurdie, P.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Bisanz, J.E. qiime2R-Importing QIIME2 Artifacts and Associated Data into R Sessions. Version 0.99.13. Available online: https://rdrr.io/github/jbisanz/qiime2R/ (accessed on 4 February 2020).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-2. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 December 2019).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: New York, NY, USA, 2016; p. 260. [Google Scholar]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. Available online: https://cran.r-project.org/package=ggpubr (accessed on 11 July 2019).
- Chen, H.; Boutros, P.C. Venndiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Chiuve, S.E.; Rexrode, K.M.; Spiegelman, D.; Logroscino, G.; Manson, J.E.; Rimm, E.B. Primary prevention of stroke by healthy lifestyle. Circulation 2008, 6, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Chiuve, S.E.; McCullough, M.L.; Sacks, F.M.; Rimm, E.B. Healthy lifestyle factors in the primary prevention of coronary heart disease among men: Benefits among users and nonusers of lipid-lowering and antihypertensive medications. Circulation 2006, 114, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.; Liu, S.; Solomon, C.G.; Willett, W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med. 2001, 345, 790–797. [Google Scholar] [CrossRef]
- Platz, E.A.; Willett, W.C.; Colditz, G.A.; Rimm, E.B.; Spiegelman, D.; Giovannucci, E. Proportion of colon cancer risk that might be preventable in a cohort of middle-aged US men. Cancer Causes Control 2000, 11, 579–588. [Google Scholar] [CrossRef]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Witkiewicz, W. Correlations of host genetics and gut microbiome composition. Front. Microbiol. 2016, 7, 1357. [Google Scholar] [CrossRef]
- Small, C.M.; Milligan-Myhre, K.; Bassham, S.; Guillemin, K.; Cresko, W.A. Host genotype and microbiota contribute asymmetrically to transcriptional variation in the threespine stickleback gut. Genome Biol. Evol. 2017, 9, 504–520. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Möller, S.; Wang, J.; Künzel, S.; Zillikens, D.; Baines, J.F.; Ibrahim, S.M. Genome-wide mapping of gene–microbiota interactions in susceptibility to autoimmune skin blistering. Nat. Commun. 2013, 4, 2462. [Google Scholar] [CrossRef] [PubMed]
- Hufeldt, M.R.; Nielsen, D.S.; Vogensen, F.K.; Midtvedt, T.; Hansen, A.K. Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comp. Med. 2010, 60, 336–347. [Google Scholar] [PubMed]
- Turroni, F.; Marchesi, J.R.; Foroni, E.; Gueimonde, M.; Shanahan, F.; Margolles, A.; van Sinderen, D.; Ventura, M. Microbiomic analysis of the bifidobacterial population in the human distal gut. ISME J. 2009, 3, 745–751. [Google Scholar] [CrossRef]
- Rerksuppaphol, S.; Rerksuppaphol, L. A randomized double-blind controlled trial of Lactobacillus acidophilus plus Bifidobacterium bifidum versus placebo in patients with hypercholesterolemia. J. Clin. Diagn. Res. 2015, 9, KC01–KC04. [Google Scholar] [CrossRef]
- Sharma, P.; Bhardwaj, P.; Singh, R. Administration of Lactobacillus casei and Bifidobacterium bifidum ameliorated hyperglycemia, dyslipidemia, and oxidative stress in diabetic rats. Int. J. Prev. Med. 2016, 7, 102. [Google Scholar]
- Lomax, A.R.; Calder, P.C. Probiotics, immune function, infection and inflammation: A review of the evidence from studies conducted in humans. Curr. Pharm. Des. 2009, 15, 1428–1518. [Google Scholar] [CrossRef]
- Salminen, S.; Nybom, S.; Meriluoto, J.; Collado, M.C.; Vesterlund, S.; El-Nezami, H. Interaction of probiotics and pathogens—Benefits to human health? Curr. Opin. Biotechnol. 2010, 21, 157–167. [Google Scholar] [CrossRef]
- Aizawa, E.; Tsuji, H.; Asahara, T.; Takahashi, T.; Teraishi, T.; Yoshida, S.; Ota, M.; Koga, N.; Hattori, K.; Kunugi, H. Possible association of Bifidobacterium and Lactobacillus in the gut microbiota of patients with major depressive disorder. J. Affect. Disord. 2016, 202, 254–257. [Google Scholar] [CrossRef]
- Queipo-Ortuno, M.I.; Seoane, L.M.; Murri, M.; Pardo, M.; Gomez-Zumaquero, J.M.; Cardona, F.; Casanueva, F.; Tinahones, F.J. Gut microbiota composition in male rat models under different nutritional status and physical activity and its association with serum leptin and ghrelin levels. PLoS ONE 2013, 8, e65465. [Google Scholar] [CrossRef]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen 2018, 8, e00678. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Xu, X.; Li, Y.; Li, X.; Yang, X.; Chen, H.; Zhu, Y.; Lu, N.; He, C. Sex-specific association between the gut microbiome and high-fat diet-induced metabolic disorders in mice. Biol. Sex Differ. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jones-hall, Y.L.; Kozik, A.; Nakatsu, C. Ablation of tumor necrosis factor is associated with decreased inflammation and alterations of the microbiota in a mouse model of inflammatory bowel disease. PLoS ONE 2015, 10, e0119441. [Google Scholar]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.D.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genom. 2014, 15, 1066. [Google Scholar] [CrossRef]
- Crost, E.H.; Tailford, L.E.; Le Gall, G.; Fons, M.; Henrissat, B.; Juge, N. Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS ONE 2013, 8, e76341. [Google Scholar] [CrossRef]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef]
- Sartor, R.B. Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 2011, 4, 127–132. [Google Scholar] [CrossRef]
- Chua, H.H.; Chou, H.C.; Tung, Y.L.; Chiang, B.L.; Liao, C.C.; Liu, H.H.; Ni, Y.H. Intestinal dysbiosis featuring abundance of Ruminococcus gnavus associates with allergic diseases in infants. Gastroenterology 2018, 154, 154–167. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Increased abundance of Sutterella spp. And Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Auchtung, T.A.; Ajami, N.J.; Velasquez, K.; Smith, D.P.; Ii, R.D.L.G.; Salas, R.; Petrosino, J.F. Effects of tobacco smoke and electronic cigarette vapor exposure on the oral and gut microbiota in humans: A pilot study. PeerJ. 2018, 6, e4693. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Allais, L.; Kerckhof, F.M.; Verschuere, S.; Bracke, K.R.; De Smet, R.; Laukens, D.; Van den Abbeele, P.; De Vos, M.; Boon, N.; Brusselle, G.G.; et al. Chronic cigarette smoke exposure induces microbial and inflammatory shifts and mucin changes in the murine gut. Environ. Microbiol. 2016, 18, 1352–1363. [Google Scholar] [CrossRef]
- Zijlstra, F.J.; Srivastava, E.D.; Rhodes, M.; van Dijk, A.P.; Fogg, F.; Samson, H.J.; Copeman, M.; Russell, M.A.; Feyerabend, C.; Williams, G.T.; et al. Effect of nicotine on rectal mucus and mucosal eicosanoids. Gut 1994, 35, 247–251. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.N.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Savin, Z.; Kivity, S.; Yonath, H.; Yehuda, S. Smoking and the intestinal microbiome. Arch. Microbiol. 2018, 200, 677–684. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef]
- Yutin, N.; Galperin, M.Y. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 2013, 15, 2631–2641. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, G.; Guo, Y.; Zhao, Y.; Zhao, M.; Lai, Y.; Sui, P.; Shi, T.; Guo, W.; Huang, Z. Variations in oral microbiota composition are associated with a risk of throat cancer. Front. Cell Infect. Microbiol. 2019, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.; Charlton, K.; Stefoska-Needham, A.; Ibrahim, R.; Lambert, K. The gut microbiota profile of adults with kidney disease and kidney stones: A systematic review of the literature. BMC Nephrol. 2020, 21, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Drago, L.; Toscano, M.; Rodighiero, V.; De Vecchi, E.; Mogna, G. Cultivable and pyrosequenced fecal microflora in centenarians and young subjects. J. Clin. Gastroenterol. 2012, 46, S81–S84. [Google Scholar] [CrossRef]
- Bruggemann, H.; Baumer, S.; Fricke, W.F.; Wiezer, A.; Liesegang, H.; Decker, I.; Herzberg, C.; Martinez-Arias, R.; Merkl, R.; Henne, A.; et al. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc. Natl. Acad. Sci. USA 2003, 100, 1316–1321. [Google Scholar] [CrossRef]
- Boer, C.G.; Radjabzadeh, D.; Medina-Gomez, C.; Garmaeva, S.; Schiphof, D.; Arp, P.; Koet, T.; Kurilshikov, A.; Fu, J.; Ikram, M.A.; et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat. Commun. 2019, 10, 4881. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Henderson, G.; Yilmaz, P.; Kumar, S.; Forster, R.J.; Kelly, W.J.; Leahy, S.C.; Guan, L.L.; Janssen, P.H. Improved taxonomic assignment of rumen bacterial 16S rRNA sequences using a revised SILVA taxonomic framework. PeerJ 2019, 7, e6496. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, J.X.; Hu, N.; Ren, J.; Du, M.; Zhu, M.J. Side-stream smoking reduces intestinal inflammation and increases expression of tight junction proteins. World J. Gastroenterol. 2012, 18, 2180–2187. [Google Scholar] [CrossRef]
- Bennett, J.E.; Dolin, R.; Blaser, M.J. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2015. [Google Scholar]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef]
- Ulger-Toprak, N.; Liu, C.; Summanen, P.H.; Finegold, S.M. Murdochiella asaccharolytica gen. nov., sp. nov., a Gram-stain-positive, anaerobic coccus isolated from human wound specimens. Int. J. Syst. Evol. Micr. 2010, 60, 1013–1016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flygel, T.T.; Sovershaeva, E.; Claassen-Weitz, S.; Hjerde, E.; Mwaikono, K.S.; Odland, J.Ø.; Ferrand, R.A.; Mchugh, G.; Gutteberg, T.J.; Nicol, M.P.; et al. Composition of gut microbiota of children and adolescents with perinatal human immunodeficiency virus infection taking antiretroviral therapy in Zimbabwe. J. Infect. Dis. 2020, 221, 483–492. [Google Scholar] [PubMed]
- Flynn, K.J.; Ruffin, M.T., 4th; Turgeon, D.K.; Schloss, P.D. Spatial variation of the native colon microbiota in healthy adults. Cancer Prev. Res. 2018, 11, 393–402. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Modifiable Lifestyle Factors | Civilians (n = 38) | Navy Trainees (n = 66) |
|---|---|---|
| Cigarette smoking 1 n (%) | ||
| Non-smokers (Never smoked) | 27 (71.05) | 47 (71.21) |
| Smokers (Currently smokes less than 1 pack per day) | 11 (28.95) | 14 (21.21) |
| Alcohol consumption 1 n (%) | ||
| Nondrinkers (Drinking frequency is less than weekly) | 18 (47.37) | 28 (42.42) |
| Regular drinkers (Drinks at least once per week) | 18 (47.37) | 23 (34.85) |
| Sleep duration 1 n (%) | ||
| 7–9 h sleep (Recommended hour) | 19 (50.00) | 52 (78.79) |
| <7 h sleep (Below recommended hour) | 19 (50.00) | 9 (13.64) |
| Physical exercise 1, n (%) | ||
| Regular exercisers (≥2 h per week) | 10 (26.32) | 26 (39.39) |
| Non exercisers (<1 h per week) | 28 (73.68) | 35 (53.03) |
| Body mass index (BMI), n (%) | ||
| <18.50 | 2 (5.26) | 1 (1.52) |
| 18.50–24.99 | 26 (68.42) | 38 (57.58) |
| 25.00–29.99 | 10 (26.32) | 22 (33.33) |
| Variables | Civilians (n = 38) | Navy Trainees (n = 66) | ||||
|---|---|---|---|---|---|---|
| T0 | T8 | p-Value | T0 | T8 | p-Value | |
| Cigarette smoking (cigarettes smoked per day) | 4.50 ± 7.88 | 3.90 ± 7.15 | 0.250 | 2.28 ± 4.73 | 0.00 ± 0.00 | <0.001 |
| Alcohol consumption (alcohol consumption days per week) | 0.83 ± 1.08 | 0.83 ± 1.08 | >0.999 | 0.90 ± 1.33 | 0.00 ± 0.00 | <0.001 |
| Sleeping duration (hours per day) | 6.37 ± 1.30 | 6.42 ± 1.18 | 0.856 | 7.69 ± 1.29 | 7.57 ± 0.49 | 0.400 |
| Physical exercise (exercise duration in the past week, min) | 84.34 ± 171.20 | 85.26 ± 155.00 | 0.587 | 178.60 ± 228.20 | 1347.00 ± 203.80 | <0.001 |
| Body mass index (BMI, kg/m2) | 22.91 ± 2.97 | 22.83 ± 2.92 | 0.632 | 24.52 ± 2.59 | 23.82 ± 2.24 | <0.001 |
| Groups | PERMANOVA | |
|---|---|---|
| Pseudo-F | p-Value | |
| All participants | 4.281 | 0.001 |
| All participants at T0 vs. T8 | 0.789 | 0.771 |
| Civilians | 5.120 | 0.001 |
| Civilians at T0 vs. T8 | 0.250 | 1.000 |
| Navy trainees | 3.674 | 0.001 |
| Navy trainees at T0 vs. T8 | 1.206 | 0.189 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.; Tagele, S.B.; Son, H.; Ibal, J.C.; Kerfahi, D.; Yun, H.; Lee, B.; Park, C.Y.; Kim, E.S.; Kim, S.-J.; et al. Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change. Microorganisms 2020, 8, 1265. https://doi.org/10.3390/microorganisms8091265
Jung Y, Tagele SB, Son H, Ibal JC, Kerfahi D, Yun H, Lee B, Park CY, Kim ES, Kim S-J, et al. Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change. Microorganisms. 2020; 8(9):1265. https://doi.org/10.3390/microorganisms8091265
Chicago/Turabian StyleJung, YeonGyun, Setu Bazie Tagele, HyunWoo Son, Jerald Conrad Ibal, Dorsaf Kerfahi, Hyunju Yun, Bora Lee, Clara Yongjoo Park, Eun Soo Kim, Sang-Jun Kim, and et al. 2020. "Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change" Microorganisms 8, no. 9: 1265. https://doi.org/10.3390/microorganisms8091265
APA StyleJung, Y., Tagele, S. B., Son, H., Ibal, J. C., Kerfahi, D., Yun, H., Lee, B., Park, C. Y., Kim, E. S., Kim, S.-J., & Shin, J.-H. (2020). Modulation of Gut Microbiota in Korean Navy Trainees following a Healthy Lifestyle Change. Microorganisms, 8(9), 1265. https://doi.org/10.3390/microorganisms8091265