Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease


Abstract
1. Introduction
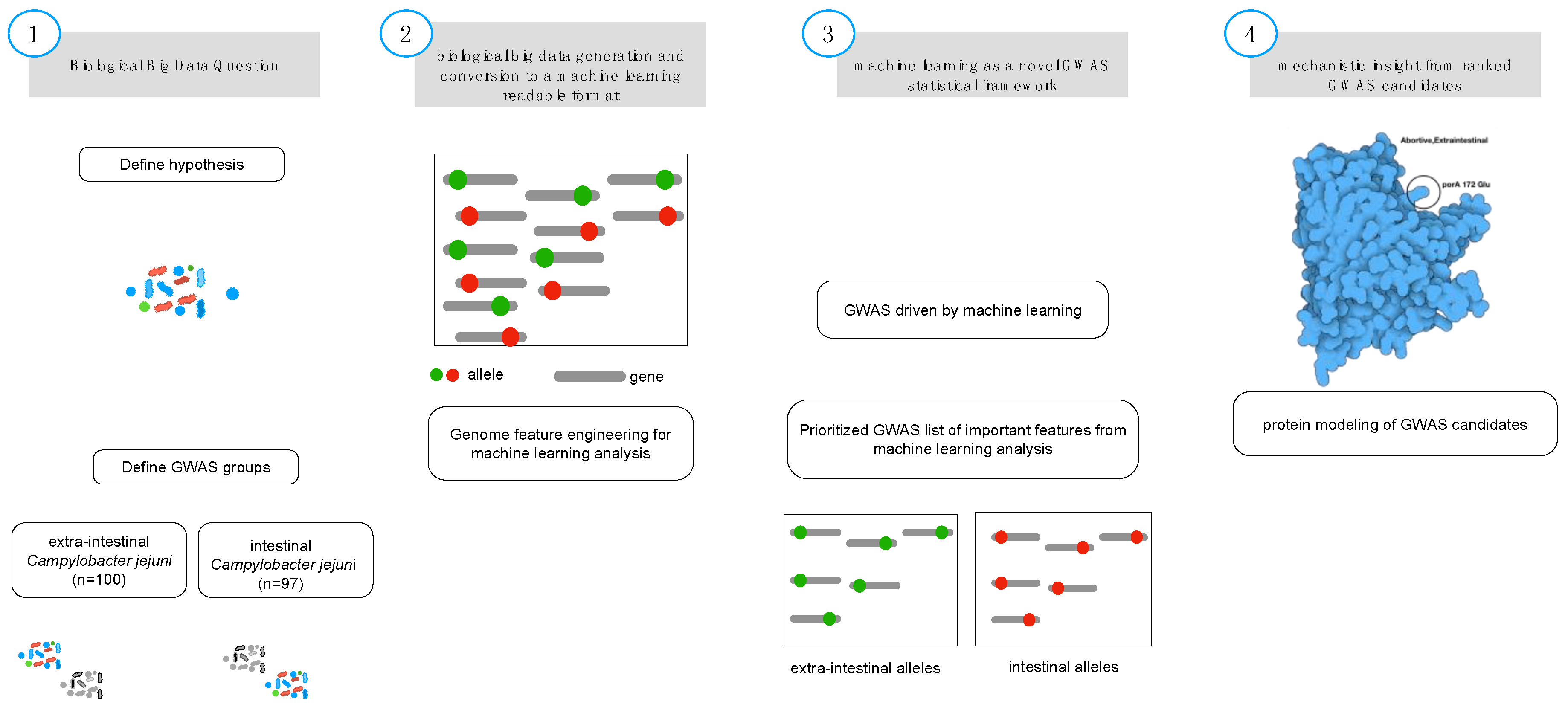
2. Materials and Methods
2.1. Biological Feature Engineering
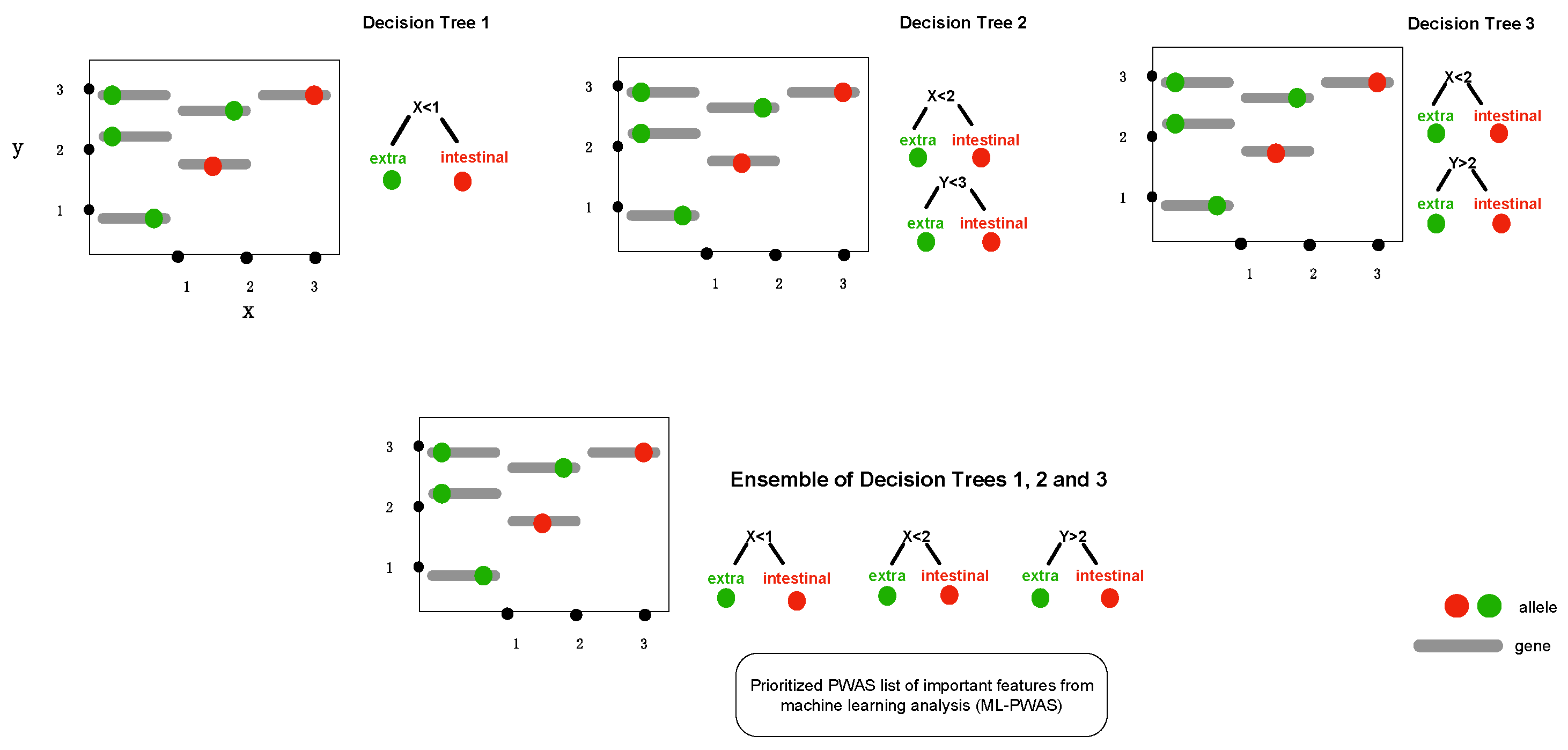
2.2. Gradient Tree Boosting as GWAS Framework
2.3. Tetris Plot
2.4. Population-Wide Whole-Genome Phylogeny
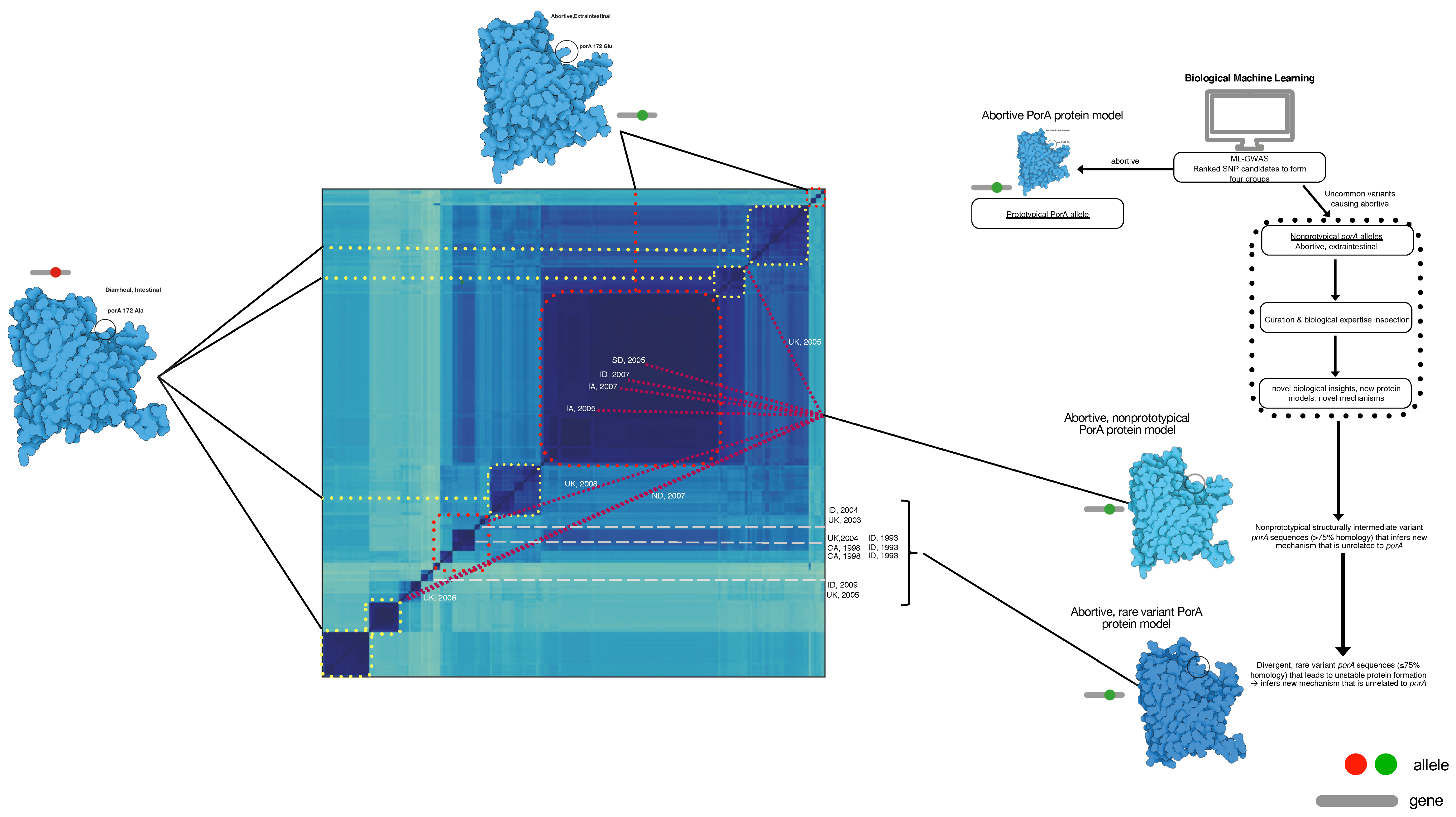
2.5. Protein Modeling
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Page, A.; Cummins, C.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Power, R.; Parkhill, J.; De Oliveira, T. Microbial genome-wide association studies: lessons from human GWAS. Nat. Rev. Genet. 2016, 18, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Nelson, G.W.; Troyer, J.L.; A Lautenberger, J.; Kessing, B.; Winkler, C.A.; O’Brien, S.J. Accounting for multiple comparisons in a genome-wide association study (GWAS). BMC Genomics 2010, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Weimer, B.C.; Storey, D.B.; Elkins, C.A.; Baker, R.C.; Markwell, P.; Chambliss, D.D.; Edlund, S.B.; Kaufman, J. Defining the food microbiome for authentication, safety, and process management. IBM J. Res. Dev. 2016, 60, 1:1–1:13. [Google Scholar] [CrossRef]
- Kaufman, J.H.; Elkins, C.A.; Davis, M.; Weis, A.M.; Huang, B.C.; Mammel, M.K.; Patel, I.R.; Beck, K.L.; Edlund, S.; Chambliss, D.; et al. Insular Microbiogeography: Three Pathogens as Exemplars. Curr. Issues Mol. Boil. 2019, 36, 89–108. [Google Scholar]
- Kaufman, J.H.; Seabolt, E.; Kunitomi, M.; Agarwal, A.; Beck, K.; Krishnareddy, H.; Weimer, B.C. Exploiting Functional Context in Biology: Reconsidering Classification of Bacterial Life. 2018 IEEE 34th Int. Conf. Data Eng. Workshops (ICDEW) 2018, 2018, 17–20. [Google Scholar]
- Kaufman, J.H.; Christopher, A.E.; Matthew, D.; Allison, M.W.; Bihua, C.H.; Mark, K.M.; Isha, R.; Patel, K.L.; Beck, S.E.; David, C.; et al. Microbiogeography and microbial genome evolution. arXiv 2017, arXiv:1703.07454. [Google Scholar]
- Breiman, L. Statistical Modeling: The Two Cultures (with comments and a rejoinder by the author). Stat. Sci. 2001, 16, 199–231. [Google Scholar] [CrossRef]
- Draper, J.; Hansen, L.M.; Bernick, D.L.; Abedrabbo, S.; Underwood, J.G.; Kong, N.; Huang, B.C.; Weis, A.; Weimer, B.C.; Van Vliet, A.H.M.; et al. Fallacy of the Unique Genome: Sequence Diversity within Single Helicobacter pylori Strains. mBio 2017, 8, e02321-16. [Google Scholar] [CrossRef]
- Heithoff, D.M.; Shimp, W.R.; House, J.K.; Xie, Y.; Weimer, B.C.; Sinsheimer, R.L.; Mahan, M.J. Intraspecies Variation in the Emergence of Hyperinfectious Bacterial Strains in Nature. PLoS Pathog. 2012, 8, e1002647. [Google Scholar] [CrossRef]
- Read, T.D.; Massey, R.C. Characterizing the genetic basis of bacterial phenotypes using genome-wide association studies: a new direction for bacteriology. Genome Med. 2014, 6, 1063. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.J.; Friedman, J.; Cordero, O.X.; Preheim, S.P.; Timberlake, S.C.; Szabó, G.; Polz, M.F.; Alm, E.J. Population Genomics of Early Events in the Ecological Differentiation of Bacteria. Science 2012, 336, 48–51. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.-R.; Nookaew, I.; Leuze, M.R.; Ahn, T.-H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.M.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.-M.; Ochman, H. The Evolution of Bacterial Genome Architecture. Front. Genet. 2017, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Carranza, E.; Barajas, H.; Alcaraz, L.D.; Servin-Gonzalez, L.; Ponce-Soto, G.Y. Variability of Bacterial Essential Genes Among Closely Related Bacteria: The Case of Escherichia coli. Front. Microbiol. 2018, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Periaswamy, B.; Sahin, O.; Yaeger, M.; Plummer, P.; Zhai, W.; Shen, Z.; Dai, L.; Chen, S.L.; Zhang, Q. Point mutations in the major outer membrane protein drive hypervirulence of a rapidly expanding clone of Campylobacter jejuni. Proc. Natl. Acad. Sci. USA 2016, 113, 10690–10695. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Bandoy, D.D. Pangenome guided pharmacophore modelling of enterohemorrhagic Escherichia coli sdiA. F1000Research 2019, 8, 33. [Google Scholar] [CrossRef]
- Brown, C.T.; Irber, L. sourmash: a library for MinHash sketching of DNA. J. Open Source Softw. 2016, 1, 27. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; De Beer, T.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2016, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Weis, A.; Clothier, K.A.; Huang, B.C.; Kong, N.; Weimer, B.C. Draft Genome Sequences of Campylobacter jejuni Strains That Cause Abortion in Livestock. Genome Announc. 2016, 4, e01324-16. [Google Scholar] [CrossRef] [PubMed]
- Weis, A.; Storey, D.B.; Taff, C.C.; Townsend, A.K.; Huang, B.C.; Kong, N.T.; Clothier, K.A.; Spinner, A.; Byrne, B.A.; Weimer, B.C. Genomic Comparison of Campylobacter spp. and Their Potential for Zoonotic Transmission between Birds, Primates, and Livestock. Appl. Environ. Microbiol. 2016, 82, 7165–7175. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining - KDD, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery (ACM): New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Behravan, H.; Hartikainen, J.M.; Tengström, M.; Pylkäs, K.; Winqvist, R.; Kosma, V.; Mannermaa, A. Machine learning identifies interacting genetic variants contributing to breast cancer risk: A case study in Finnish cases and controls. Sci. Rep. 2018, 8, 13149. [Google Scholar] [CrossRef]
- Yang, J.; Wright, S.N.; Hamblin, M.; McCloskey, U.; Alcantar, M.A.; Schrübbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661.e9. [Google Scholar] [CrossRef]
- Nilsson, I.; Von Heijne, G. Fine-tuning the topology of a polytopic membrane protein: Role of positively and negatively charged amino acids. Cell 1990, 62, 1135–1141. [Google Scholar] [CrossRef]
- Elazar, A.; Weinstein, J.J.; Prilusky, J.; Fleishman, S. Interplay between hydrophobicity and the positive-inside rule in determining membrane-protein topology. Proc. Natl. Acad. Sci. USA 2016, 113, 10340–10345. [Google Scholar] [CrossRef]
- Kim, C.; Schmidt, T.; Cho, E.-G.; Ye, F.; Ulmer, T.S.; Ginsberg, M.H. Basic amino-acid side chains regulate transmembrane integrin signalling. Nature 2011, 481, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Adler, B.; Haridas, S.; Albert, M.J. PorA protein of Campylobacter jejuni is not a cytotoxin mediating inflammatory diarrhoea. Microbes Infect. 2005, 7, 853–859. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GWAS Statistical Metrics | Machine Learning Coupled to GWAS Metrics | |||
|---|---|---|---|---|
| Allele | GWAS p-Value | Bonferroni Corrected p-Value | Candidate Ranking | Feature Importance |
| X1 | 0.001 | 8.3 × 10−10 | 1 | 80 |
| X2 | 0.001 | 8.3 × 10−10 | 2 | 75 |
| X3 | 0.001 | 8.3 × 10−10 | 3 | 70 |
| Xn | 0.001 | 8.3 × 10−10 | Rankn | Importancen |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandoy, D.D.R.; Weimer, B.C. Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms 2020, 8, 549. https://doi.org/10.3390/microorganisms8040549
Bandoy DDR, Weimer BC. Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms. 2020; 8(4):549. https://doi.org/10.3390/microorganisms8040549
Chicago/Turabian StyleBandoy, DJ Darwin R., and Bart C. Weimer. 2020. "Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease" Microorganisms 8, no. 4: 549. https://doi.org/10.3390/microorganisms8040549
APA StyleBandoy, D. D. R., & Weimer, B. C. (2020). Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms, 8(4), 549. https://doi.org/10.3390/microorganisms8040549