Long-Term Rewetting of Three Formerly Drained Peatlands Drives Congruent Compositional Changes in Pro- and Eukaryotic Soil Microbiomes through Environmental Filtering

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
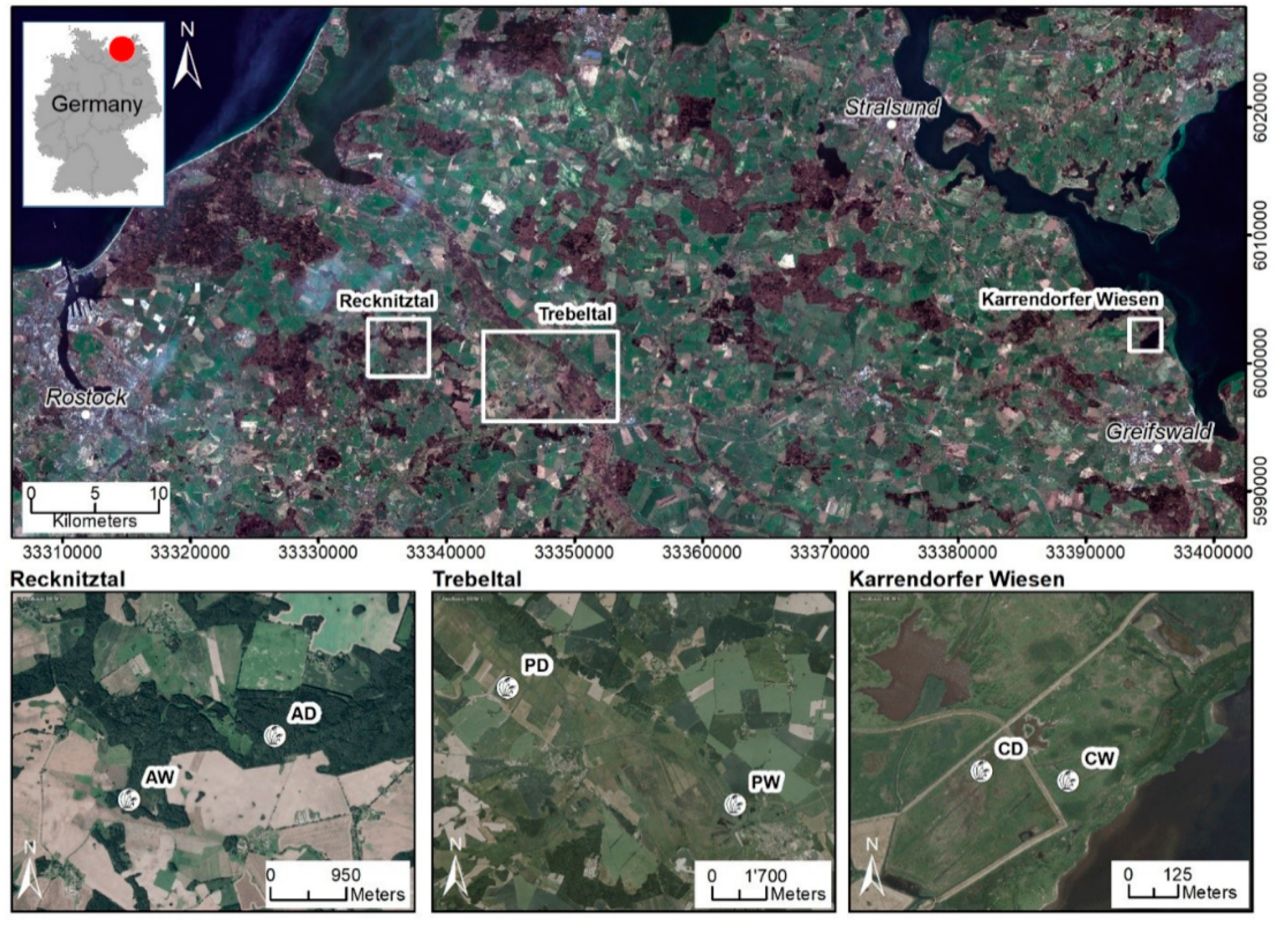
2.1. Sites and Sampling
2.2. Soil Physicochemical Properties
2.3. DNA Extraction and PCR/qPCR
2.4. Amplicon Sequencing and Bioinformatics
2.5. Gas Flux Measurements
3. Results
3.1. Study Sites and Soil Properties
3.2. Microbiome Analysis
3.2.1. Diversity of Prokaryotes and Eukaryotes
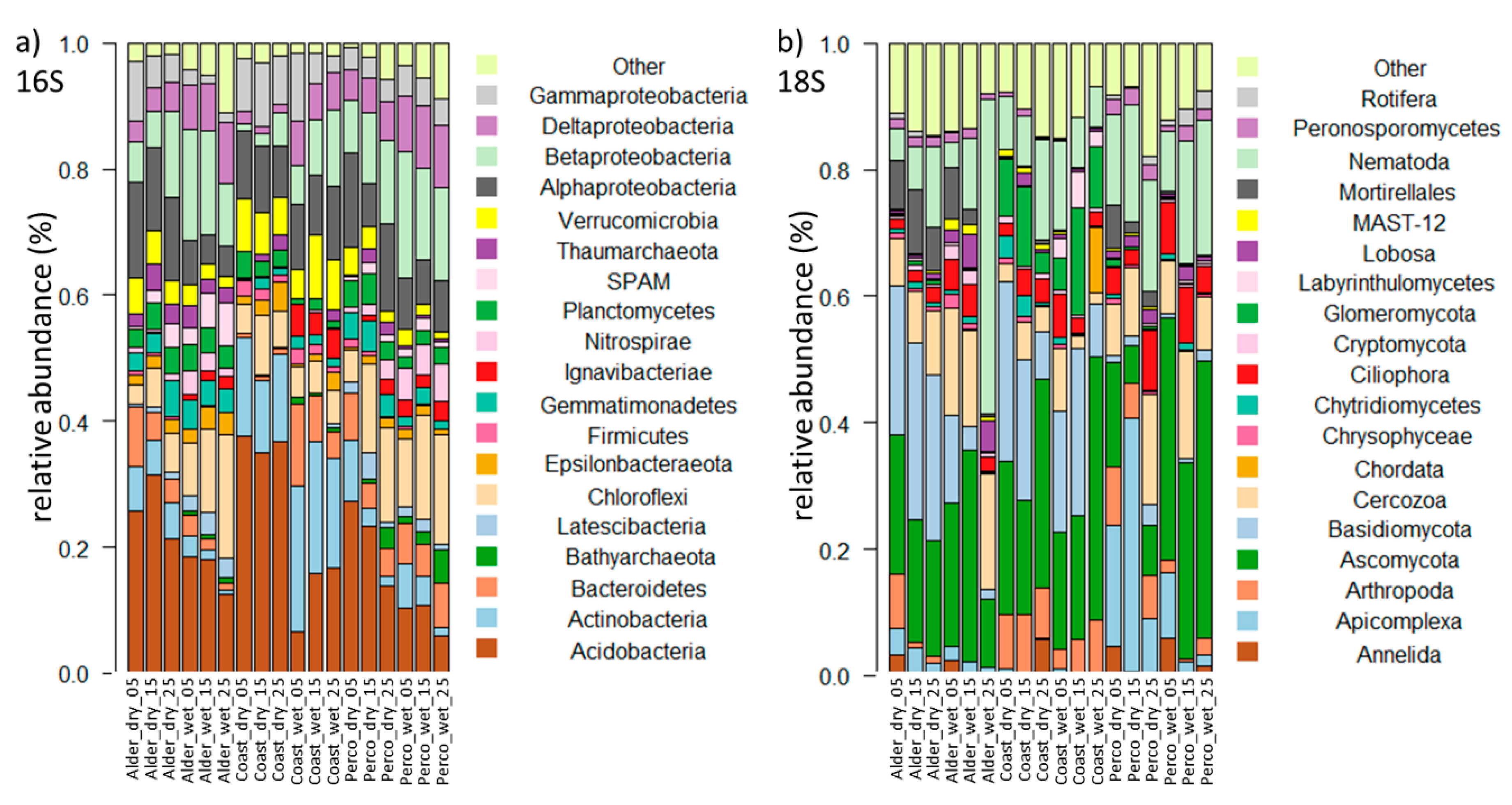
3.2.2. Microbiota Composition
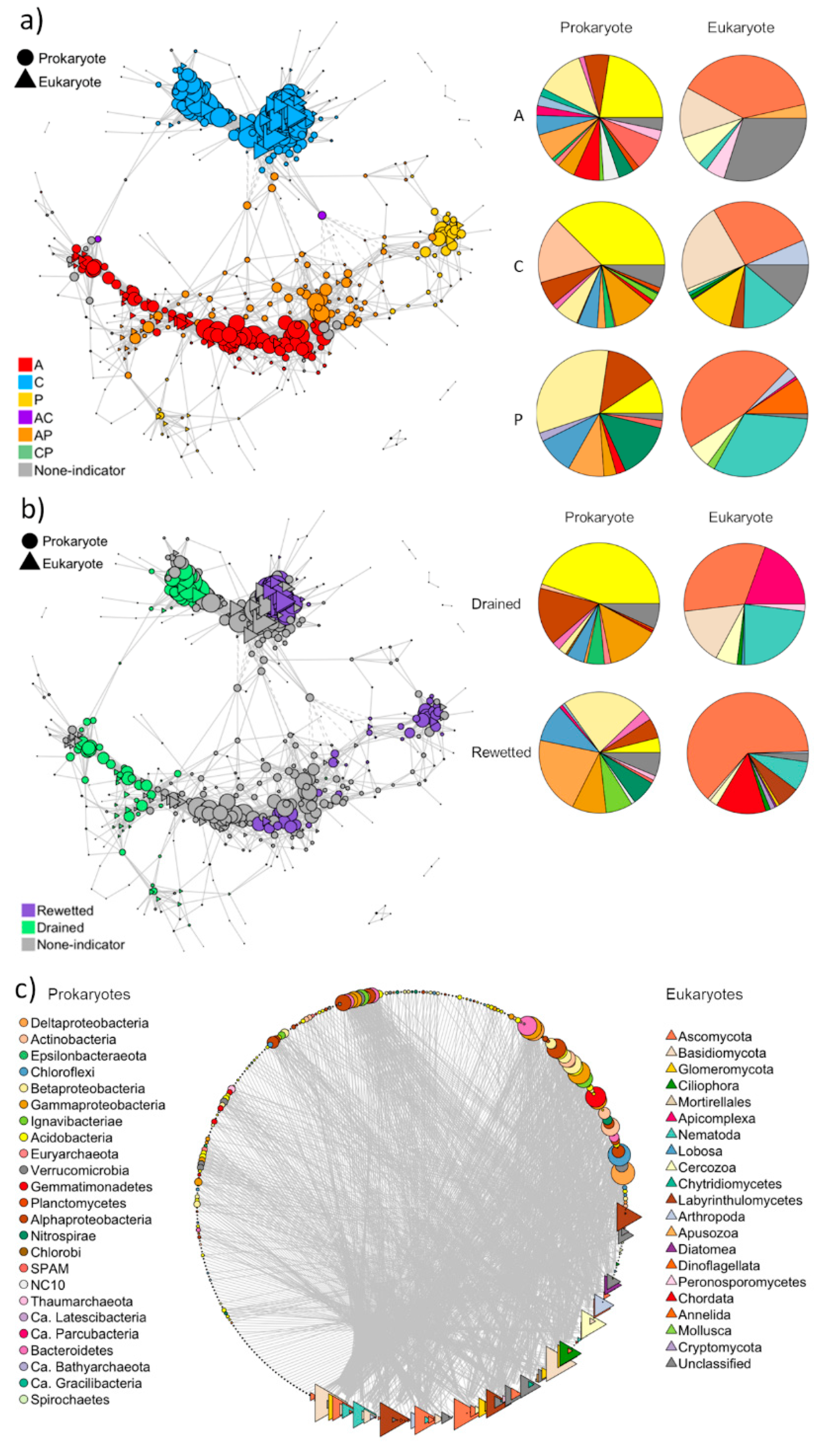
3.2.3. Co-Occurrence Network Analysis
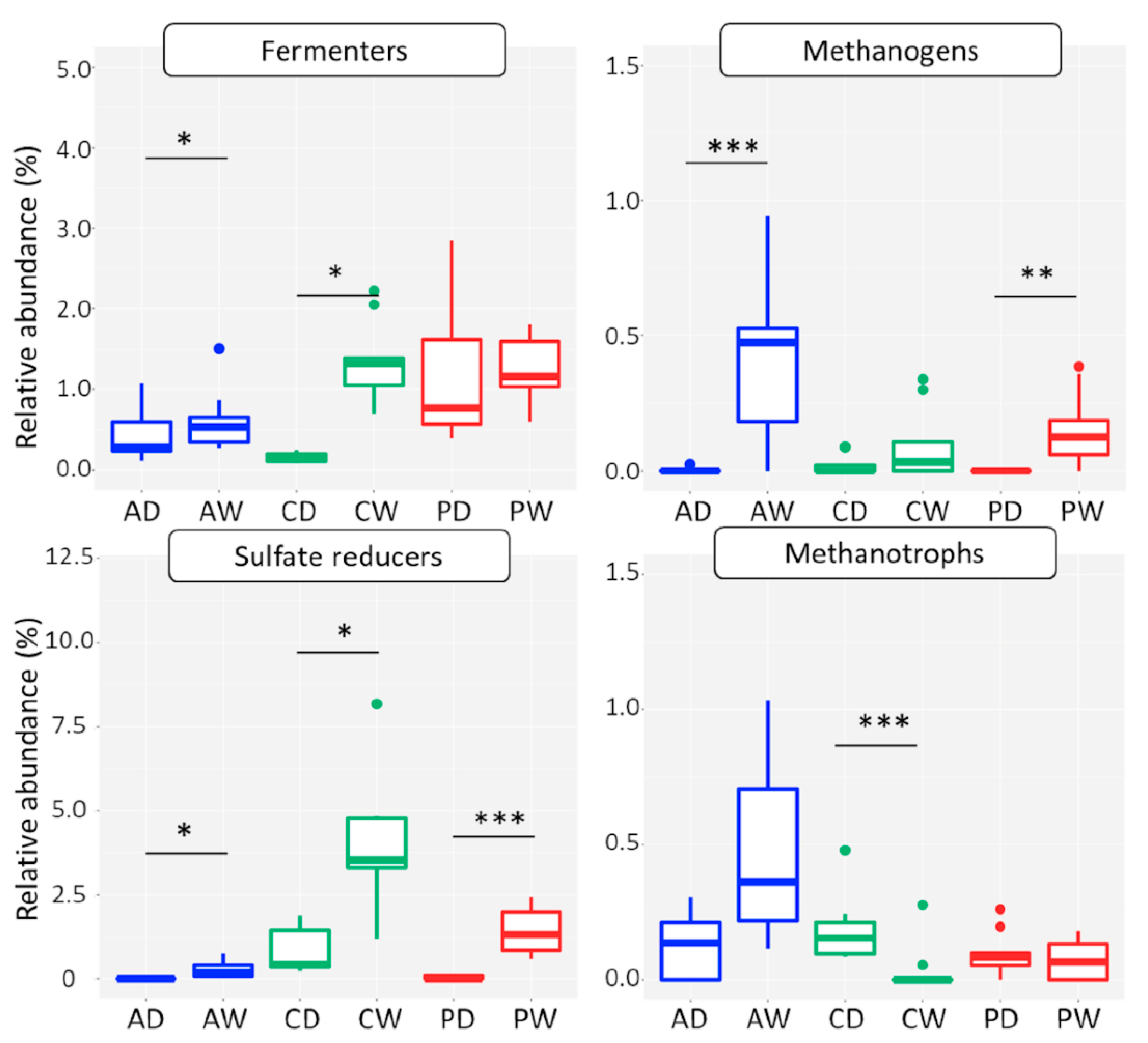
3.2.4. Functional Groups
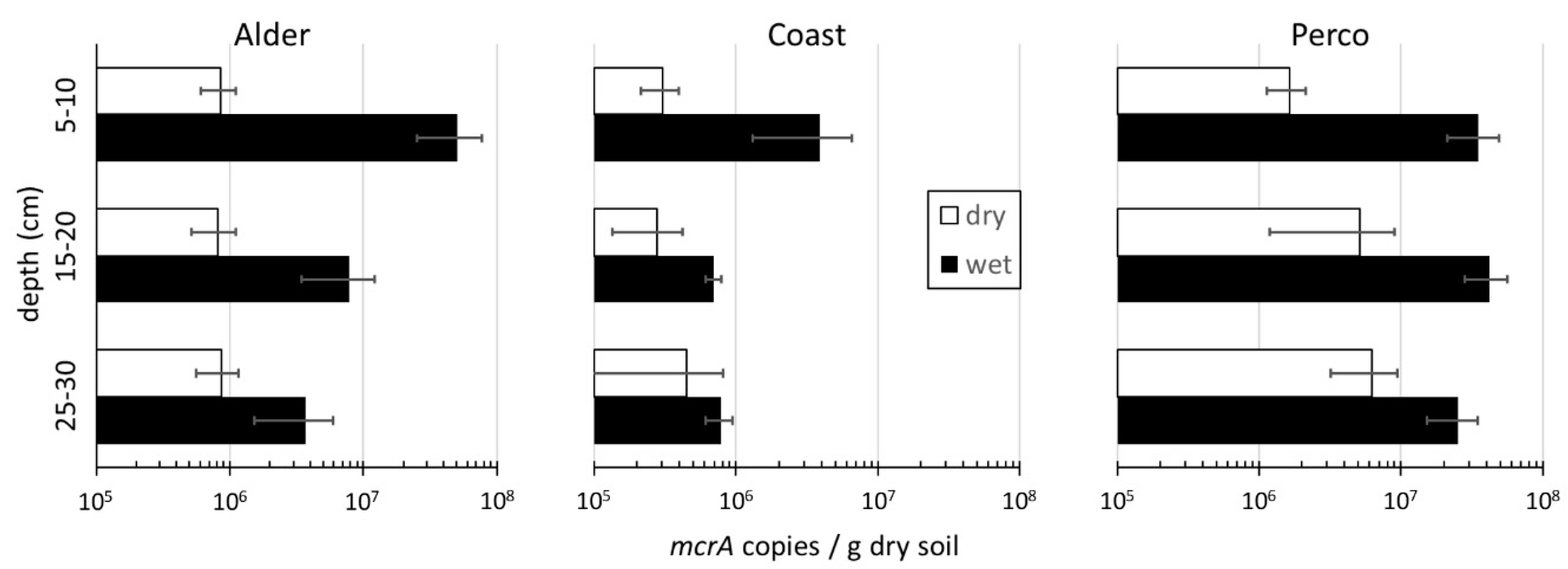
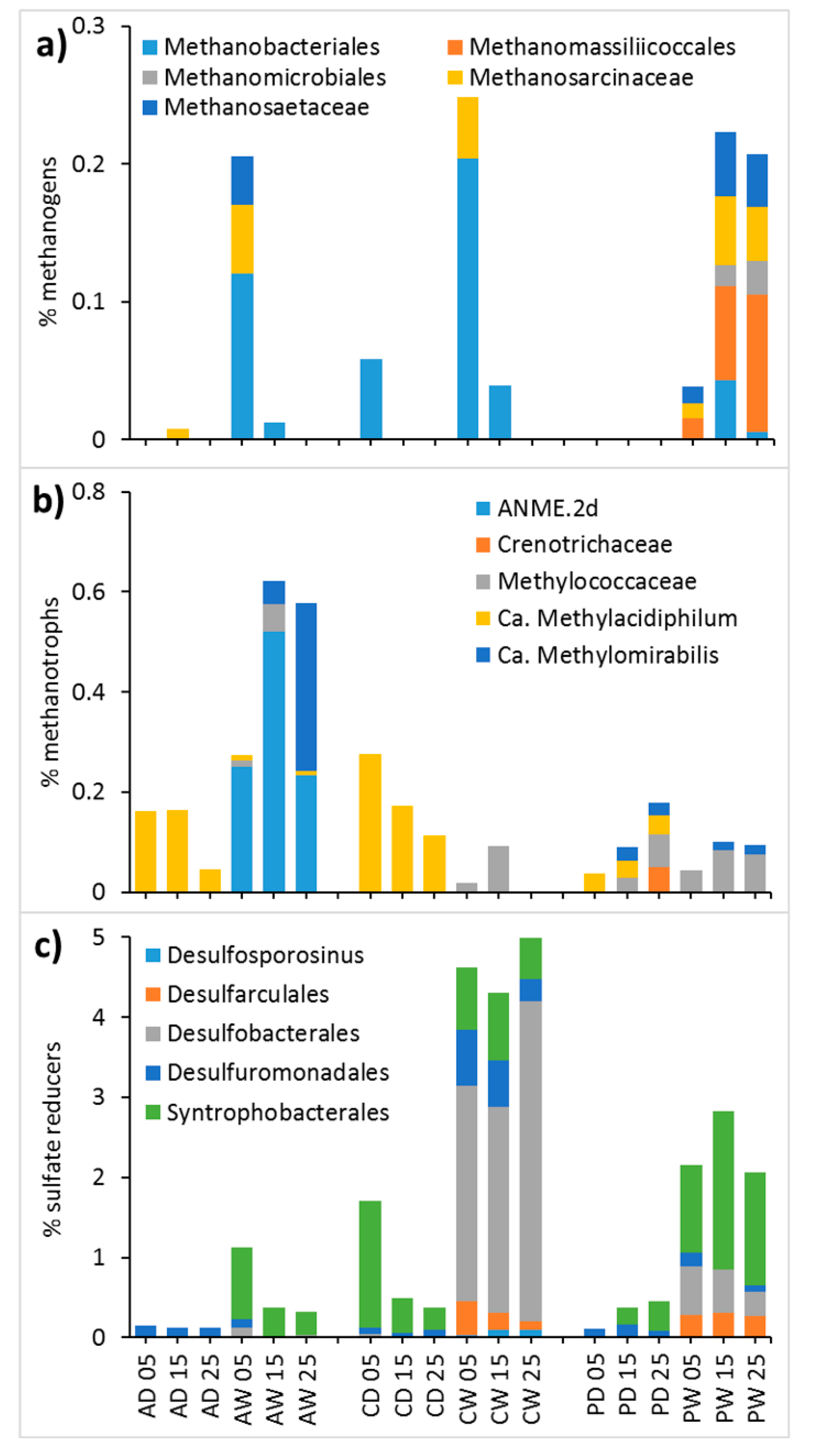
Methanogens
Methanotrophs
Sulfate Reducers
3.3. Greenhouse Gas Emissions
4. Discussion
4.1. Environmental Filtering Effects on Pro- and Eukaryotic Community Composition
4.2. The Influence of Rewetting on Methanogens
4.3. Interactions between Methanogens, Methanotrophs, and Sulfate Reducers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Joosten, H.; Sirin, A.; Couwenberg, J.; Laine, J.; Smith, P. The role of peatlands in climate regulation. In Peatland Restoration and Ecosystem Services: Science, Policy and Practice; Bonn, A., Allott, T., Evans, M., Joosten, H., Stoneman, R., Eds.; Cambridge University Press: Cambridge, UK, 2016; pp. 63–76. [Google Scholar]
- Freibauer, A.; Drösler, M.; Gensior, A.; Schulze, E.D. Das Potenzial von Wäldern und Mooren für den Klimaschutz in Deutschland und auf globaler Ebene. Nat. Landsch 2009, 84, 20–25. [Google Scholar]
- Joosten, H.; Couwenberg, J. Are Emission Reductions from Peatlands Measurable, Reportable and Verifiable? Wetlands International: Wageningen, The Netherlands, 2009; Volume 14. [Google Scholar]
- Ziebarth, M.; Lenschow, U.; Permien, T. Konzept zum Schutz und zur Nutzung der Moore—Fortschreibung des Konzeptes zur Bestandssicherung und zur Entwicklung der Moore; Umwelt und Verbraucherschutz Mecklenburg-Vorpommern: Schwerin, Germany, 2009; Volume 109. [Google Scholar]
- Greifswald Mire Center. Moorbodenschutz als naturbasierte Lösung im Klimaschutzprogramm 2030—schnelle Einstellung von Moor-Entwässerung für wirkungsvollen Klimaschutz nötig! Greifswald Mire Center: Greifswald, Germany, 2019. [Google Scholar]
- Dean, J.F.; Middelburg, J.J.; Röckmann, T.; Aerts, R.; Blauw, L.G.; Egger, M.; Jetten, M.S.M.; de Jong, A.E.E.; Meisel, O.H.; Rasigraf, O.; et al. Methane Feedbacks to the Global Climate System in a Warmer World. Rev. Geophys. 2018, 56, 207–250. [Google Scholar] [CrossRef]
- Parish, F.; Sirin, A.; Charman, D.; Joosten, H.; Minayeva, T.; Silvius, M.; Stringer, L. Assessment on Peatlands, Biodiversity and Climate Change: Main Report; Global Environment Centre, Kuala Lumpur & Wetlands International: Wageningen, The Netherlands, 2008; ISBN 978-983-43751-0-2. [Google Scholar]
- Conrad, R. Soil Microorganisms as Controllers of Atmospheric Trace Gases (H2, CO, CH4, OCS, N2O, and NO). Microbiol. Mol. Biol. Rev. 1996, 60, 609–640. [Google Scholar] [CrossRef]
- Andersen, R.; Chapman, S.J.; Artz, R.R.E. Microbial communities in natural and disturbed peatlands: A review. Soil Biol. Biochem. 2013, 57, 979–994. [Google Scholar] [CrossRef]
- Chimner, R.A.; Cooper, D.J.; Wurster, F.C.; Rochefort, L. An overview of peatland restoration in North America: Where are we after 25 years? Restor. Ecol. 2017, 25, 283–292. [Google Scholar] [CrossRef]
- Couwenberg, J.; Thiele, A.; Tanneberger, F.; Augustin, J.; Bärisch, S.; Dubovik, D.; Liashchynskaya, N.; Michaelis, D.; Minke, M.; Skuratovich, A.; et al. Assessing greenhouse gas emissions from peatlands using vegetation as a proxy. Hydrobiologia 2011, 674, 67–89. [Google Scholar] [CrossRef]
- Hahn, J.; Köhler, S.; Glatzel, S.; Jurasinski, G. Methane exchange in a coastal fen in the first year after flooding—A systems shift. PLoS ONE 2015, 10, e0140657. [Google Scholar] [CrossRef]
- Estop-Aragonés, C.; Zajac, K.; Blodau, C. Effects of extreme experimental drought and rewetting on CO2 and CH4 exchange in mesocosms of 14 European peatlands with different nitrogen and sulfur deposition. Glob. Chang. Biol. 2016, 22, 2285–2300. [Google Scholar] [CrossRef]
- Wilson, D.; Blain, D.; Cowenberg, J.; Evans, C.D.; Murdiyarso, D.; Page, S.E.; Renou-Wilson, F.; Rieley, J.O.; Sirin, A.; Strack., M.; et al. Greenhouse gas emission factors associated with rewetting of organic soils. Mires Peat 2016, 17, 1–28. [Google Scholar]
- Lyu, Z.; Shao, N.; Akinyemi, T.; Whitman, W.B. Methanogenesis. Curr. Biol. 2018, 28, R727–R732. [Google Scholar] [CrossRef]
- Wen, X.; Unger, V.; Jurasinski, G.; Koebsch, F.; Horn, F.; Rehder, G.; Sachs, T.; Zak, D.; Lischeid, G.; Knorr, K.H.; et al. Predominance of methanogens over methanotrophs in rewetted fens characterized by high methane emissions. Biogeosciences 2018, 15, 6519–6536. [Google Scholar] [CrossRef]
- Yarwood, S.A. The role of wetland microorganisms in plant-litter decomposition and soil organic matter formation: A critical review. FEMS Microbiol. Ecol. 2018, 94, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tveit, A.T.; Urich, T.; Frenzel, P.; Svenning, M.M. Metabolic and trophic interactions modulate methane production by Arctic peat microbiota in response to warming. Proc. Natl. Acad. Sci. USA 2015, 112, E2507–E2516. [Google Scholar] [CrossRef] [PubMed]
- Söllinger, A.; Schwab, C.; Weinmaier, T.; Loy, A.; Tveit, A.T.; Schleper, C.; Urich, T. Phylogenetic and genomic analysis of Methanomassiliicoccales in wetlands and animal intestinal tracts reveals clade-specific habitat. FEMS Microbiol. Ecol. 2016, 92, 1–12. [Google Scholar] [CrossRef]
- Welte, C.U.; Rasigraf, O.; Vaksmaa, A.; Versantvoort, W.; Arshad, A.; Op den Camp, H.J.M.; Jetten, M.S.M.; L�ke, C.; Reimann, J.; Lüke, C.; et al. Nitrate- and nitrite-dependent anaerobic oxidation of methane. Environ. Microbiol. Rep. 2016, 8, 941–955. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Makarova, K.S.; Abbas, B.; Ferrer, M.; Golyshin, P.N.; Galinski, E.A.; Ciordia, S.; Mena, M.C.; Merkel, A.Y.; Wolf, Y.I.; et al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2017, 2, 17081. [Google Scholar] [CrossRef]
- Singleton, C.; McCalley, C.; Woodcroft, B.; Boyd, J.; Evans, P.; Hodgkins, S.; Chanton, J.; Frolking, S.; Crill, P.; Saleska, S.; et al. Methanotrophy across a natural permafrost thaw environment. ISME J. 2018, 1, 2544–2558. [Google Scholar] [CrossRef]
- Bonn, A.; Allott, T.; Evans, M.; Joosten, H.; Stoneman, R. Peatland Restoration and Ecosystem Services: Science, Policy and Practice; Cambridge University Press: Cambridge, UK, 2016; ISBN 9781139177788. [Google Scholar]
- Cadotte, M.W.; Tucker, C.M. Should Environmental Filtering be Abandoned? Trends Ecol. Evol. 2017, 32, 429–437. [Google Scholar] [CrossRef]
- Huber, S.A.; Balz, A.; Abert, M.; Pronk, W. Characterisation of aquatic humic and non-humic matter with size-exclusion chromatography—Organic carbon detection—Organic nitrogen detection (LC-OCD-OND). Water Res. 2011, 45, 879–885. [Google Scholar] [CrossRef]
- Heinz, M.; Zak, D. Storage effects on quantity and composition of dissolved organic carbon and nitrogen of lake water, leaf leachate and peat soil water. Water Res. 2018, 130, 98–104. [Google Scholar] [CrossRef]
- Steinberg, L.M.; Regan, J.M. mcrA-targeted real-time quantitative PCR method to examine methanogen communities. Appl. Environ. Microbiol. 2009, 75, 4435–4442. [Google Scholar] [CrossRef]
- Angel, R.; Matthies, D.; Conrad, R.; Meyer, A.; Kilian, E. Activation of Methanogenesis in Arid Biological Soil Crusts Despite the Presence of Oxygen. PLoS ONE 2011, 6, e20453. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K. Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009-15. [Google Scholar]
- Ray, J.L.; Althammer, J.; Skaar, K.S.; Simonelli, P.; Larsen, A.; Stoecker, D.; Sazhin, A.; Ijaz, U.Z.; Quince, C.; Nejstgaard, J.C.; et al. Metabarcoding and metabolome analyses of copepod grazing reveal feeding preference and linkage to metabolite classes in dynamic microbial plankton communities. Mol. Ecol. 2016, 25, 5585–5602. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 17 December 2019).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Lanzén, A.; Jørgensen, S.L.; Huson, D.H.; Gorfer, M.; Grindhaug, S.H.; Jonassen, I.; Øvreås, L.; Urich, T. CREST—Classification Resources for Environmental Sequence Tags. PLoS ONE 2012, 7, e49334. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, 1–12. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R package version 2.5-6. Available online: http://cran.r-project.org/package=vegan (accessed on 17 December 2019).
- Harrell, F.E.; Dunpont, C. Hmisc: Harrell Miscellaneous. R package version 4.2-0. Available online: https://CRAN.R-project.org/package=Hmisc (accessed on 17 December 2019).
- Louca, S.; Wegener Parfrey, L.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Csardi, G. Nepusz T: The Igraph Software Package for Complex Network Research, InterJournal, Complex Systems 1695. Available online: http://igraph.org (accessed on 17 December 2019).
- Livingston, G.P.; Hutchinson, G.L. Enclosure-based measurement of trace gas exchange: Applications and sources of error. In Biogenic Trace Gases: Measuring Emissions from Soil and Water; Matson, P.A., Harris, R.C., Eds.; Wiley: Hoboken, NJ, USA, 1995; pp. 14–53. [Google Scholar]
- Günther, A.; Schenzle, E.; Jurasinski, G. Flexible Chambers for Greenhouse Gas Measurements in Tall Vegetation. Tech. Rep. 2014. [Google Scholar] [CrossRef]
- De Cáceres, M.; Legendre, P. Associations between species and groups of sites: Indices and statistical inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997, 61, 262–280. [Google Scholar] [CrossRef]
- He, S.; Malfatti, S.A.; Mcfarland, J.W.; Anderson, F.E.; Pati, A.; Huntemann, M.; Tremblay, J.; Glavina, T.; Waldrop, M.P.; Windham-myers, L.; et al. Patterns in Wetland Microbial Community Composition and Functional Gene Repertoire Associated with Methane Emissions. Mbio 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lamers, L.P.M.; van Roozendaal, S.M.E.; Roelofs, J.G.M. Acidification of Freshwater Wetlands: Combined Effects of Non-Airborne Sulfur Pollution and Desiccation. Water. Air. Soil Pollut. 1998, 105, 95–106. [Google Scholar] [CrossRef]
- Cusell, C.; Lamers, L.P.M.; van Wirdum, G.; Kooijman, A. Impacts of water level fluctuation on mesotrophic rich fens: Acidification vs. eutrophication. J. Appl. Ecol. 2013, 50, 998–1009. [Google Scholar] [CrossRef]
- Eichorst, S.A.; Trojan, D.; Roux, S.; Herbold, C.; Rattei, T.; Woebken, D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 2018, 20, 1041–1063. [Google Scholar] [CrossRef]
- Hausmann, B.; Pelikan, C.; Herbold, C.W.; Köstlbacher, S.; Albertsen, M.; Eichorst, S.A.; Glavina Del Rio, T.; Huemer, M.; Nielsen, P.H.; Rattei, T.; et al. Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 2018, 12, 1729–1742. [Google Scholar] [CrossRef]
- Dang, C.; Morrissey, E.M.; Virginia, W.; Neubauer, S.C.; Franklin, R.B. Novel microbial community composition and carbon biogeochemistry emerge over time following saltwater intrusion in wetlands. Glob. Chang. Biol. 2018, 25, 549–561. [Google Scholar] [CrossRef]
- Nazaries, L.; Murrell, J.C.; Millard, P.; Baggs, L.; Singh, B.K. Methane, microbes and models: Fundamental understanding of the soil methane cycle for future predictions. Environ. Microbiol. 2013, 15, 2395–2417. [Google Scholar] [CrossRef] [PubMed]
- Reim, A.; Hernández, M.; Klose, M.; Chidthaisong, A.; Yuttitham, M.; Conrad, R. Response of methanogenic microbial communities to desiccation stress in flooded and rain-fed paddy soil from Thailand. Front. Microbiol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Schöfl, M.; Zak, D.; Minke, M.; Gelbrecht, J.; Augustin, J.; Freibauer, A. Organic sediment formed during inundation of a degraded fen grassland emits large fluxes of CH4 and CO2. Biogeosciences 2011, 8, 1539–1550. [Google Scholar] [CrossRef]
- Angle, J.C.; Morin, T.H.; Solden, L.M.; Narrowe, A.B.; Smith, G.J.; Borton, M.A.; Rey-Sanchez, C.; Daly, R.A.; Mirfenderesgi, G.; Hoyt, D.W.; et al. Methanogenesis in oxygenated soils is a substantial fraction of wetland methane emissions. Nat. Commun. 2017, 8, 1567. [Google Scholar] [CrossRef] [PubMed]
- Harpenslager, S.F.; van den Elzen, E.; Kox, M.A.R.; Smolders, A.J.P.; Ettwig, K.F.; Lamers, L.P.M. Rewetting former agricultural peatlands: Topsoil removal as a prerequisite to avoid strong nutrient and greenhouse gas emissions. Ecol. Eng. 2015, 84, 159–168. [Google Scholar] [CrossRef]
- Zak, D.; Goldhammer, T.; Cabezas, A.; Gelbrecht, J.; Gurke, R.; Wagner, C.; Reuter, H.; Augustin, J.; Klimkowska, A.; McInnes, R. Top soil removal reduces water pollution from phosphorus and dissolved organic matter and lowers methane emissions from rewetted peatlands. J. Appl. Ecol. 2018, 55, 311–320. [Google Scholar] [CrossRef]
- Neubauer, S.C.; Franklin, R.B.; Berrier, D.J. Saltwater intrusion into tidal freshwater marshes alters the biogeochemical processing of organic carbon. Biogeosciences 2013, 10, 8171–8183. [Google Scholar] [CrossRef]
- van Dijk, G.; Nijp, J.J.; Metselaar, K.; Lamers, L.P.M.; Smolders, A.J.P. Salinity-induced increase of the hydraulic conductivity in the hyporheic zone of coastal wetlands. Hydrol. Process. 2017, 31, 880–890. [Google Scholar] [CrossRef]
- Mihajlovski, A.; Doré, J.; Levenez, F.; Alric, M.; Brugère, J.F. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2010, 2, 272–280. [Google Scholar] [CrossRef]
- Zalman, C.A.; Meade, N.; Chanton, J.; Kostka, J.E.; Bridgham, S.D.; Keller, J.K. Methylotrophic methanogenesis in Sphagnum-dominated peatland soils. Soil Biol. Biochem. 2017, 118, 156–160. [Google Scholar] [CrossRef]
- Knief, C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 2015, 6, 1346. [Google Scholar] [CrossRef] [PubMed]
- van Teeseling, M.C.F.; Pol, A.; Harhangi, H.R.; van der Zwart, S.; Jetten, M.S.M.; Op den Camp, H.J.M.; van Niftrik, L. Expanding the verrucomicrobial methanotrophic world: Description of three novel species of Methylacidimicrobium gen. nov. Appl. Environ. Microbiol. 2014, 80, 6782–6791. [Google Scholar] [CrossRef] [PubMed]
- Vaksmaa, A.; Lüke, C.; van Alen, T.; Valè, G.; Lupotto, E.; Jetten, M.S.M.; Ettwig, K.F. Distribution and activity of the anaerobic methanotrophic community in a nitrogen-fertilized Italian paddy soil. FEMS Microbiol. Ecol. 2016, 92, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tveit, A.T.; Urich, T.; Svenning, M.M. Metatranscriptomic analysis of arctic peat soil microbiota. Appl. Environ. Microbiol. 2014, 80, 5761–5772. [Google Scholar] [CrossRef]
- Kristjansson, J.K.; Schönheit, P. Why do sulfate-reducing bacteria outcompete methanogenic bacteria for substrates? Oecologia 1983, 60, 264–266. [Google Scholar] [CrossRef]
- Thauer, R.K.; Kaster, A.-K.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef]
- Kristjansson, J.K.; Schönheit, P.; Thauer, R.K. Different Ks values for hydrogen of methanogenic bacteria and sulfate reducing bacteria: An explanation for the apparent inhibition of methanogenesis by sulfate. Arch. Microbiol. 1982, 131, 278–282. [Google Scholar] [CrossRef]
- Poffenbarger, H.J.; Needelman, B.A.; Megonigal, J.P. Salinity influence on methane emissions from tidal marshes. Wetlands 2011, 31, 831–842. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbr. | Fen Type | State (Year of Rewetting) | Water Table | Peat Thickness | Dominant Plant Species |
|---|---|---|---|---|---|
| Alder | Alder carr | dry | −100 cm | 60 cm | Black Alder (Alnus glutinosa), Ground Elder (Aegopodium podagraria), and Common Nettle (Urtica dioica) |
| wet (1999) | +15 cm | >1 m | Black Alder (Alnus glutinosa), Greater Pond Sedge (Carex riparia) | ||
| Coast | Coastal fen | dry | −70 cm | 70 cm | Creeping Bentgrass (Agrostis stolonifera) |
| wet (1993) | −5 cm | 30 cm | Creeping Bentgrass (Agrostis stolonifera) | ||
| Perco | Percolation fen | dry | −20 cm | 6 m | Creeping Buttercup (Ranunculus repens), Tufted Hairgrass (Deschampsia cespitosa) |
| wet (1998) | +10 cm | 6 m | Lesser Pond Sedge (Carex acutiformis) |
| Site | CH4 Flux (mg m−2 h−1) | Ecosystem Respiration CO2 Flux (mg m−2 h−1) | mcrA_DW (Copies g−1 Soil Dry Weight) April 2017 | mcrA_DW (Copies g−1 Soil Dry Weight) August 2017 | ||||
|---|---|---|---|---|---|---|---|---|
| Alder_dry | −0.08 | ± 0.07 | 493 | ± 406 | 8.5 × 105 | ± 2.9 × 105 | 3.14 × 105 | ±3.30 × 104 |
| Alder_wet | 0.88 * | ± 0.51 | 47 † | ± 70 | 2.1 × 107 | ± 2.6 × 107 | 3.89 × 107 | ±1.66 × 106 |
| Coast_dry | −0.11 | ± 0.14 | 2024 | ± 585 | 3.5 × 105 | ± 2.5 × 105 | 3.62 × 105 | ±9.97 × 104 |
| Coast_wet | 0.00 * | ± 0.01 | 1420 | ± 311 | 1.8 × 106 | ± 2.1 × 106 | 5.32 × 105 | ±6.90 × 104 |
| Perco_dry | −0.12 | ± 0.05 | 783 | ± 152 | 4.3 × 106 | ± 3.5 × 106 | 6.31 × 106 | ±1.07 × 106 |
| Perco_wet | 15.8 * | ± 23.8 | 550 † | ± 232 | 3.4 × 107 | ± 1.5 × 107 | 5.28 × 107 | ±3.55 × 106 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weil, M.; Wang, H.; Bengtsson, M.; Köhn, D.; Günther, A.; Jurasinski, G.; Couwenberg, J.; Negassa, W.; Zak, D.; Urich, T. Long-Term Rewetting of Three Formerly Drained Peatlands Drives Congruent Compositional Changes in Pro- and Eukaryotic Soil Microbiomes through Environmental Filtering. Microorganisms 2020, 8, 550. https://doi.org/10.3390/microorganisms8040550
Weil M, Wang H, Bengtsson M, Köhn D, Günther A, Jurasinski G, Couwenberg J, Negassa W, Zak D, Urich T. Long-Term Rewetting of Three Formerly Drained Peatlands Drives Congruent Compositional Changes in Pro- and Eukaryotic Soil Microbiomes through Environmental Filtering. Microorganisms. 2020; 8(4):550. https://doi.org/10.3390/microorganisms8040550
Chicago/Turabian StyleWeil, Micha, Haitao Wang, Mia Bengtsson, Daniel Köhn, Anke Günther, Gerald Jurasinski, John Couwenberg, Wakene Negassa, Dominik Zak, and Tim Urich. 2020. "Long-Term Rewetting of Three Formerly Drained Peatlands Drives Congruent Compositional Changes in Pro- and Eukaryotic Soil Microbiomes through Environmental Filtering" Microorganisms 8, no. 4: 550. https://doi.org/10.3390/microorganisms8040550
APA StyleWeil, M., Wang, H., Bengtsson, M., Köhn, D., Günther, A., Jurasinski, G., Couwenberg, J., Negassa, W., Zak, D., & Urich, T. (2020). Long-Term Rewetting of Three Formerly Drained Peatlands Drives Congruent Compositional Changes in Pro- and Eukaryotic Soil Microbiomes through Environmental Filtering. Microorganisms, 8(4), 550. https://doi.org/10.3390/microorganisms8040550