Metagenomics-Based Proficiency Test of Smoked Salmon Spiked with a Mock Community

 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Laboratory Workflows of Participants
2.3. Data Sharing
2.4. Bioinformatics and Statistical Analysis
3. Results
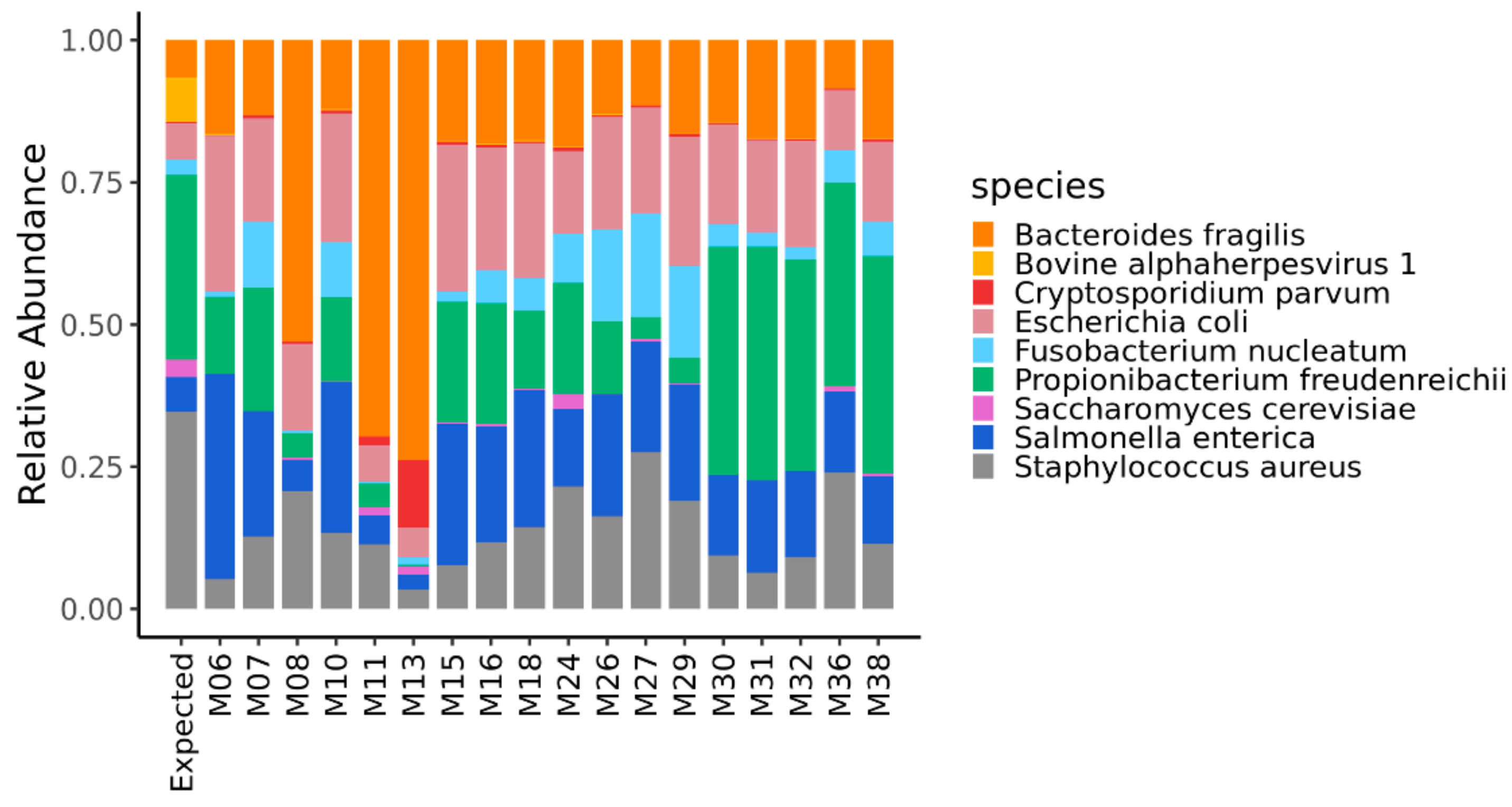
3.1. Relative Abundance of the Reads Assigned to the Taxonomic Domains and the Microorganisms of the Mock Community
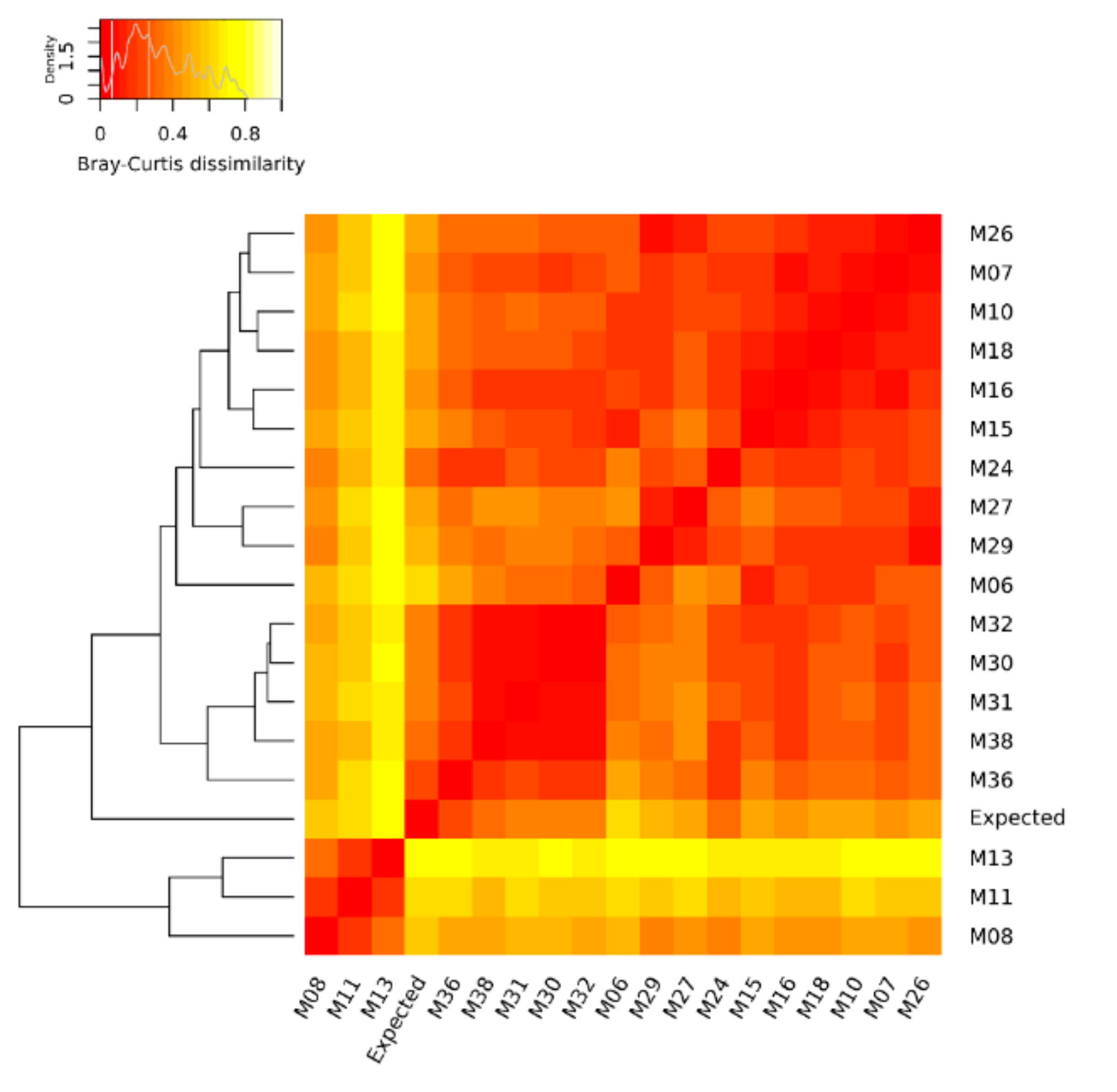
3.2. Ranking of the Metagenomic Datasets Based on Their Dissimilarity to the Expected Composition and Assessment of the Impact of Each Variable of the Workflow on the Abundance of the Mock Community Members
3.3. Assessment of the Impact of Each Workflow on the Abundance of the Mock Community Members
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority (EFSA). Scientific report on the European Union One Health 2018 Zoonoses Report. EFSA J. 2019, 17, 5926. [Google Scholar]
- Yang, X.; Noyes, N.R.; Doster, E.; Martin, J.N.; Linke, L.M.; Magnuson, R.J.; Hua, Y.; Ifigenia, G.; Dale, R.W.; Kenneth, L.J.; et al. Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within the microbiome of the beef production chain. Appl. Environ. Microbiol. 2016, 82, 2433–2443. [Google Scholar] [CrossRef]
- Larsen, M.H.; Dalmasso, M.; Ingmer, H.; Langsrud, S.; Malakauskas, M.; Mader, A.; Trond Møretrø, T.; Sonja Smole Mozina, S.S.; Rychli, K.; Wagner, M.; et al. Persistence of foodborne pathogens and their control in primary and secondary food production chains. Food Cont. 2014, 44, 92–109. [Google Scholar] [CrossRef]
- Aw, T.G.; Wengert, S.; Rose, J.B. Metagenomic analysis of viruses associated with field-grown and retail lettuce identifies human and animal viruses. Int. J. Food Microbiol. 2016, 223, 50–56. [Google Scholar] [CrossRef]
- Leonard, S.R.; Mammel, M.K.; Lacher, D.W.; Elkins, C.A. Strain-level discrimination of Shiga toxin-producing Escherichia coli in spinach using metagenomic sequencing. PLoS ONE 2016, 11, e0167870. [Google Scholar] [CrossRef] [PubMed]
- Kirstahler, P.; Bjerrum, S.S.; Friis-Møller, A.; La Cour, M.; Aarestrup, F.M.; Westh, H.; Pamp, S.J. Genomics-based identification of microorganisms in human ocular body fluid. Sci. Rep. 2018, 8, 4126. [Google Scholar] [CrossRef] [PubMed]
- Wylezich, C.; Papa, A.; Beer, M.; Höper, D. A versatile sample processing workflow for metagenomic pathogen detection. Sci. Rep. 2018, 8, 13108. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffrong, S.; Van Der Vossenh, J.; Tangi, S.; Katasej, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 2017, 5, 94. [Google Scholar] [CrossRef]
- Knudsen, B.E.; Bergmark, L.; Munk, P.; Lukjancenko, O.; Priemé, A.; Aarestrup, F.M.; Pamp, S.J. Impact of sample type and DNA isolation procedure on genomic inference of microbiome composition. MSystems 2016, 1. [Google Scholar] [CrossRef]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Segutitan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, Y.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.R.; Rawat, A.; Tang, P.; Jithesh, P.V.; Thomas, E.; Tan, R.; Tilley, P. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J. Clin. Microbiol. 2016, 54, 919–927. [Google Scholar] [CrossRef]
- Forbes, J.D.; Knox, N.C.; Ronholm, J.; Pagotto, F.; Reimer, A. Metagenomics: The next culture-independent game changer. Front. Microbiol. 2017, 8, 1069. [Google Scholar] [CrossRef]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef]
- Brinkmann, A.; Andrusch, A.; Belka, A.; Wylezich, C.; Höper, D.; Pohlmann, A.; Nordahl Petersen, t.; Lucas, P.; Blanchard, Y.; Papa, A.; et al. Proficiency testing of virus diagnostics based on bioinformatics analysis of simulated in silico high-throughput sequencing data sets. J. Clin. Microbiol. 2019, 57, e00466-19. [Google Scholar] [CrossRef]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.; Pinel, N.; May, P.; Wilmes, P. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Gen. Biol. 2016, 17, 260. [Google Scholar] [CrossRef]
- Treangen, T.J.; Koren, S.; Sommer, D.D.; Liu, B.; Astrovskaya, I.; Ondov, B.; Darling, A.; Darling, A.E.; Phillippy, A.M.; Pop, M. MetAMOS: A modular and open source metagenomic assembly and analysis pipeline. Gen. Biol. 2013, 14, R2. [Google Scholar] [CrossRef]
- Petersen, T.N.; Lukjancenko, O.; Thomsen, M.C.F.; Sperotto, M.M.; Lund, O.; Aarestrup, F.M.; Sicheritz-Pontén, T. MGmapper: Reference-based mapping and taxonomy annotation of metagenomics sequence reads. PLoS ONE 2017, 12, e0176469. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Kultima, J.R.; Coelho, L.P.; Forslund, K.; Huerta-Cepas, J.; Li, S.S.; Driessen, M.; Voigt, A.Y.; Zeller, G.; Sunagawa, S.; Bork, P. MOCAT2: A metagenomic assembly, annotation and profiling framework. Bioinformatics 2016, 32, 2520–2523. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.S.; Krumm, N.; Greenfield, N.B. One codex: A sensitive and accurate data platform for genomic microbial identification. BioRxiv 2015, 027607. [Google Scholar] [CrossRef]
- Scheuch, M.; Höper, D.; Beer, M. RIEMS: A software pipeline for sensitive and comprehensive taxonomic classification of reads from metagenomics datasets. BMC Bioinform. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Amid, C.; Pakseresht, N.; Silvester, N.; Jayathilaka, S.; Lund, O.; Dynovski, L.D.; Pataki, B.A.; Visontai, D.; Xavier, B.B.; Alako, B.T.; et al. The COMPARE Data Hubs. Database 2019, 2019, baz136. [Google Scholar] [CrossRef]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. In Microbial Environmental Genomics (MEG); Humana Press: New York, NY, USA, 2016; pp. 207–233. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucl. Acids Res. 2005, 33 (Suppl. S1), D501–D504. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Gen. Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Andersen, S.C.; Hoorfar, J. Surveillance of foodborne pathogens: Towards diagnostic metagenomics of fecal samples. Genes 2018, 9, 14. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards); Koutsoumanis, K.; Allende, A.; Alvarez-Ordonez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Hilbert, F.; et al. Scientific Opinion on the whole genome sequencing and metagenomics for outbreak investigation, source attribution and risk assessment of food-borne microorganisms. EFSA J. 2019, 17, 5898. [Google Scholar]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Ottesen, A.; Ramachandran, P.; Reed, E.; White, J.R.; Hasan, N.; Subramanian, P.; Ryan, G.; Jarvis, K.; Grim, G.; Daquiqan, N.; et al. Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol. 2016, 16, 275. [Google Scholar] [CrossRef]
- Walsh, A.M.; Crispie, F.; Daari, K.; O’Sullivan, O.; Martin, J.C.; Arthur, C.T.; Claesson, M.J.; Scott, K.P.; Cotter, P.D. Strain-level metagenomic analysis of the fermented dairy beverage nunu highlights potential food safety risks. Appl. Environ. Microbiol. 2017, 83, e01144-17. [Google Scholar] [CrossRef]
- Baym, M.; Kryazhimskiy, S.; Lieberman, T.D.; Chung, H.; Desai, M.M.; Kishony, R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 2015, 10, e0128036. [Google Scholar] [CrossRef]
- Thézé, J.; Li, T.; du Plessis, L.; Bouquet, J.; Kraemer, M.U.; Somasekar, S.; Yu, G.; de Cesare, M.; Balmaseda, A.; Kuan, G.; et al. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe 2018, 23, 855–864. [Google Scholar] [CrossRef]
- Strubbia, S.; Schaeffer, J.; Oude Munnink, B.B.; Besnard, A.; Phan, M.V.T.; Nieuwenhuijse, D.F.; de Graaf, M.; Schapendonk, C.M.E.; Wacrenier, C.; Cotton, M.; et al. Metavirome Sequencing to Evaluate Norovirus Diversity in Sewage and Related Bioaccumulated Oysters. Front. Microbiol. 2019, 10, 2394. [Google Scholar] [CrossRef]
- Peabody, M.A.; Van Rossum, T.; Lo, R.; Brinkman, F.S. Evaluation of shotgun metagenomics sequence classification methods using in silico and in vitro simulated communities. BMC Bioinform. 2015, 16, 362. [Google Scholar] [CrossRef]
- Grützke, J.; Malorny, B.; Hammerl, J.A.; Busch, A.; Tausch, S.H.; Tomaso, H.; Deneke, C. Fishing in the Soup—Pathogen Detection in Food Safety Using Metabarcoding and Metagenomic Sequencing. Front. Microbiol. 2019, 10, 1805. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Yan, Q.; Yu, Y. How much metagenomic sequencing is enough to achieve a given goal? Sci. Rep. 2013, 3, 1968. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Sanchez-Flores, A.; Baruch, M.Q. Metagenomic analysis of a Mexican ripened cheese reveals a unique complex microbiota. Food Microbiol. 2016, 57, 116–127. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Taxon (Genome Size) | Number per Subsample (Cells/Virus Genome Copies) | Expected Relative Abundance | Feature |
|---|---|---|---|
| Bacteria | |||
| Bacteroides fragilis NCTC 9343/DSM 2151 (5,241,700 bp) | 5 × 107 | 0.065 | Gram − |
| Escherichia coli ATCC 25922 (5,166,282 * bp) | 5 × 107 | 0.064 | Gram − |
| Fusobacterium nucleatum subsp. nucleatum ATCC 25586/DSM 15,643 (2,177,300 * bp) | 5 × 107 | 0.027 | Gram − |
| Propionibacterium freudenreichii subsp. Freudenreichii DSM 20271 (2,649,166 bp) | 5 × 108 | 0.331 | Gram + |
| Salmonella enterica subsp. enterica serovar Typhimurium str. ATCC 14028S/DSM 19587 (4,964,097 bp) | 5 × 107 | 0.062 | Gram − |
| Staphylococcus aureus subsp. aureus NCTC 8325 (2,821,361 bp) | 5 × 108 | 0.352 | Gram + |
| Viruses | |||
| Bovine alphaherpesvirus 1 (135,098 *) | 2.41 × 109 | <0.001 | ds DNA |
| Border disease virus isolate Gifhorn (12,325 bp) | 6 × 106 | <0.001 | ssRNA |
| Bovine viral diarrhea virus type 1 isolate NADL (12,578 bp) | 3 × 105 | <0.001 | ssRNA |
| Eukaryota | |||
| Cryptosporidium parvum IOWA II isolate (9,102,324 bp) | 1 × 106 | 0.002 | |
| Saccharomyces cerevisiae S288C (12,157,105 bp) | 5 × 106 | 0.015 |
| Metagenome Dataset | Nucleic Acid | Pre-Processing | Category Label | Extraction Kit | Category Label | cDNA Generation | Library Kit | Category Label | Sequencing Strategy | Read Length | Sequencing Platform | Category Label | Gbp | Workflow Label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M06 | DNA | BBTL | BBTL | QIAamp Fast DNA Stool | OTHER_EXD | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 1.34 | WF1 | |
| M15 | DNA | BBTL | BBTL | QIAamp Fast DNA Stool | OTHER_EXD | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 2.2 | WF1 | |
| M24 | DNA | NO_PP | NO_PP | DNeasy PowerSoil | OTHER_EXD | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 8.95 | WF2 | |
| M29 | DNA | NO_PP | NO_PP | DNeasy PowerSoil | OTHER_EXD | Nextera Flex | NexteraXT | Paired-end | 150 | MiniSeq | OTHER_SP | 3.43 | WF2 | |
| M38 | DNA | NO_PP | NO_PP | DNeasy PowerSoil | OTHER_EXD | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 12.2 | WF2 | |
| M33 | RNA | PEGO | OTHER_PP | NucliSENS MiniMag | OTHER_EXR | SS IV RT | NEBNext | OTHER_L | Paired-end | 150 | MiSeq | OTHER_SP | 0.92 | WF3 |
| M34 | RNA | PEGO | OTHER_PP | NucliSENS MiniMag | OTHER_EXR | SS IV RT | NEBNext | OTHER_L | Paired-end | 150 | MiSeq | OTHER_SP | 0.86 | WF3 |
| M16 | DNA | BBTL | BBTL | DNesasy PowerFood | PowerFood | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 1.83 | WF4 | |
| M18 | DNA | BBTL | BBTL | DNesasy PowerFood | PowerFood | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 2.50 | WF4 | |
| M23 | RNA | BBTL | BBTL | Direct-zol RNA | OTHER_EXR | cDNA SS | TruSeq | OTHER_L | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 11.01 | WF5 |
| M30 | DNA | C | OTHER_PP | DNesasy PowerFood | PowerFood | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 7.97 | WF6 | |
| M31 | DNA | C | OTHER_PP | DNesasy PowerFood | PowerFood | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 9.81 | WF6 | |
| M32 | DNA | C | OTHER_PP | DNesasy PowerFood | PowerFood | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 8.44 | WF6 | |
| M07 | DNA | NO_PP | NO_PP | QIAamp UCP Pathogen | QIAamp | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 1.03 | WF7 | |
| M10 | DNA | NO_PP | NO_PP | QIAamp UCP Pathogen | QIAamp | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 1.77 | WF7 | |
| M26 | DNA | BBTL | BBTL | QIAamp | QIAamp | TruSeq | OTHER_L | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 12.2 | WF8 | |
| M36 | DNA | BBTL | BBTL | QIAamp | QIAamp | TruSeq | OTHER_L | Paired-end | 250 | HiSeq 2500 | NextSeq 500 | 8.88 | WF8 | |
| M25 | RNA | NO_PP | NO_PP | QIAamp Viral RNA | QIAamp | cDNA SS | NexteraXT | NexteraXT | Paired-end | 150 | NextSeq 500 | NextSeq 500 | 8.64 | WF9 |
| M37 | RNA | NO_PP | NO_PP | QIAamp Viral RNA | QIAamp | SS IV RT | NexteraXT | NexteraXT | Paired-end | 200 | MiSeq | OTHER_SP | 8.84 | WF10 |
| M08 | DNA | HCFHN | OTHER_PP | QIAamp + SISPA | QIAamp | NexteraXT | NexteraXT | Paired-end | 300 | MiSeq | OTHER_SP | 1.99 | WF11 | |
| M11 | DNA | HCFHN | OTHER_PP | QIAamp + SISPA | QIAamp | NexteraXT | NexteraXT | Paired-end | 300 | MiSeq | OTHER_SP | 2.07 | WF11 | |
| M13 | DNA | HCFHN | OTHER_PP | QIAamp + SISPA | QIAamp | NexteraXT | NexteraXT | Paired-end | 300 | MiSeq | OTHER_SP | 2.62 | WF11 | |
| M27 | DNA | CP | OTHER_PP | QIAamp | QIAamp | GeneRead | OTHER_L | Single-end | 250 | Ion Torrent S5XL | OTHER_SP | 2.14 | WF12 | |
| M19 | RNA | BBTL | BBTL | RNeasy Mini kit | RNeasy Mini | cDNA SS | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 4.95 | WF13 |
| M20 | RNA | BBTL | BBTL | RNeasy Mini kit | RNeasy Mini | cDNA SS | NexteraXT | NexteraXT | Paired-end | 120 | NextSeq 500 | NextSeq 500 | 5.06 | WF13 |
| M12 | RNA | HCFHN | OTHER_PP | RNeasy Mini + SISPA | RNeasy Mini | SS IV RT | NexteraXT | NexteraXT | Paired-end | 300 | MiSeq | OTHER_SP | 2.63 | WF14 |
| M28 | RNA | CP | OTHER_PP | RNeasy Mini kit | RNeasy Mini | cDNA SS | GeneRead | OTHER_L | Single-end | 250 | Ion Torrent S5XL | OTHER_SP | 2.25 | WF15 |
| Metagenomic Dataset Label | N. Reads | % Eukaryota | % Bacteria | % Viruses | % Archaea | N. Reads Mock Community (%) |
|---|---|---|---|---|---|---|
| M06 | 1,644,354 | 14.631 | 85.004 | 0.319 | 0.025 | 875,934 (53.27) |
| M07 | 847,827 | 20.857 | 78.846 | 0.249 | 0.036 | 369,389 (43.57) |
| M08 | 742,029 | 80.338 | 19.050 | 0.325 | 0.286 | 20,440 (2.76) |
| M10 | 1,071,120 | 25.539 | 74.097 | 0.326 | 0.028 | 388,946 (36.31) |
| M11 | 730,610 | 80.567 | 18.823 | 0.309 | 0.299 | 3763 (0.52) |
| M12 | 537,347 | 79.502 | 19.747 | 0.469 | 0.264 | 11,073 (2.06) |
| M13 | 993,696 | 72.565 | 26.222 | 0.488 | 0.725 | 10,600 (1.07) |
| M15 | 1,508,927 | 21.857 | 77.846 | 0.254 | 0.033 | 635,158 (42.09) |
| M16 | 1,206,052 | 24.369 | 75.370 | 0.216 | 0.036 | 496,223 (41.14) |
| M18 | 1,867,262 | 19.308 | 80.394 | 0.259 | 0.027 | 681,708 (35.61) |
| M19 | 16,187 | 31.951 | 67.770 | 0.031 | 0.043 | 826 (5.1) |
| M20 | 16,702 | 64.459 | 35.475 | 0.048 | 0.018 | 1225 (7.33) |
| M23 | 82,614 | 45.051 | 54.713 | 0.171 | 0.036 | 21,891 (26.50) |
| M24 | 3,304,160 | 31.519 | 68.311 | 0.117 | 0.046 | 1,373,626 (41.57) |
| M25 | 1,173,758 | 45.668 | 54.015 | 0.222 | 0.073 | 353,797 (30.14) |
| M26 | 4,803,071 | 40.370 | 59.363 | 0.230 | 0.028 | 1,332,603 (27.75) |
| M27 | 911,713 | 46.621 | 53.249 | 0.104 | 0.021 | 205,421 (22.53) |
| M28 | 2340 | 72.393 | 27.564 | 0.043 | 0 | 203 (8.68) |
| M29 | 1,507,815 | 35.777 | 64.055 | 0.115 | 0.045 | 430,397 (28.54) |
| M30 | 3,680,106 | 14.271 | 85.576 | 0.122 | 0.022 | 2,217,739 (60.26) |
| M31 | 3,360,140 | 17.916 | 81.949 | 0.104 | 0.021 | 1,883,366 (56.05) |
| M32 | 4,884,497 | 11.043 | 88.809 | 0.119 | 0.020 | 2,769,020 (56.69) |
| M33 | 203,116 | 15.636 | 45.945 | 38.344 | 0.072 | 2156 (1.06) |
| M34 | 267,871 | 2.957 | 93.85 | 3.017 | 0.084 | 2231 (0.83) |
| M37 | 1,735,966 | 48.139 | 51.513 | 0.265 | 0.076 | 511,159 (29.45) |
| M36 | 17,120,850 | 4.586 | 95.202 | 0.148 | 0.057 | 9,405,164 (54.93) |
| M38 | 5,803,430 | 21.492 | 78.322 | 0.135 | 0.044 | 2,329,995 (40.15) |
| Metagenomic Dataset | Distance from the Mock Community | Rank |
|---|---|---|
| M36 | 0.207 | 1 |
| M38 | 0.337 | 2 |
| M24 | 0.345 | 3 |
| M30 | 0.361 | 4 |
| M32 | 0.370 | 5 |
| M31 | 0.395 | 6 |
| M07 | 0.435 | 7 |
| M16 | 0.447 | 8 |
| M27 | 0.464 | 9 |
| M26 | 0.488 | 10 |
| M18 | 0.495 | 11 |
| M10 | 0.496 | 12 |
| M15 | 0.498 | 13 |
| M29 | 0.544 | 14 |
| M08 | 0.556 | 15 |
| M06 | 0.610 | 16 |
| M11 | 0.646 | 17 |
| M13 | 0.790 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala, C.; Mordhorst, H.; Grützke, J.; Brinkmann, A.; Petersen, T.N.; Poulsen, C.; Cotter, P.D.; Crispie, F.; Ellis, R.J.; Castellani, G.; et al. Metagenomics-Based Proficiency Test of Smoked Salmon Spiked with a Mock Community. Microorganisms 2020, 8, 1861. https://doi.org/10.3390/microorganisms8121861
Sala C, Mordhorst H, Grützke J, Brinkmann A, Petersen TN, Poulsen C, Cotter PD, Crispie F, Ellis RJ, Castellani G, et al. Metagenomics-Based Proficiency Test of Smoked Salmon Spiked with a Mock Community. Microorganisms. 2020; 8(12):1861. https://doi.org/10.3390/microorganisms8121861
Chicago/Turabian StyleSala, Claudia, Hanne Mordhorst, Josephine Grützke, Annika Brinkmann, Thomas N. Petersen, Casper Poulsen, Paul D. Cotter, Fiona Crispie, Richard J. Ellis, Gastone Castellani, and et al. 2020. "Metagenomics-Based Proficiency Test of Smoked Salmon Spiked with a Mock Community" Microorganisms 8, no. 12: 1861. https://doi.org/10.3390/microorganisms8121861
APA StyleSala, C., Mordhorst, H., Grützke, J., Brinkmann, A., Petersen, T. N., Poulsen, C., Cotter, P. D., Crispie, F., Ellis, R. J., Castellani, G., Amid, C., Hakhverdyan, M., Guyader, S. L., Manfreda, G., Mossong, J., Nitsche, A., Ragimbeau, C., Schaeffer, J., Schlundt, J., ... De Cesare, A. (2020). Metagenomics-Based Proficiency Test of Smoked Salmon Spiked with a Mock Community. Microorganisms, 8(12), 1861. https://doi.org/10.3390/microorganisms8121861