Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis gracilipes

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Molecular Genetic Assays
2.2. Wolbachia Draft Genome and ddRAD-Seq
2.2.1. De Novo Assembly of Wolbachia Draft Genome
2.2.2. ddRAD-Seq and Wolbachia SNP Filtering
2.3. Phylogenomic Tree Reconstruction and Mitochondrial Network Analysis
2.4. Wolbachia Transcriptome Analysis
2.5. Test for Signature of Selection
3. Results
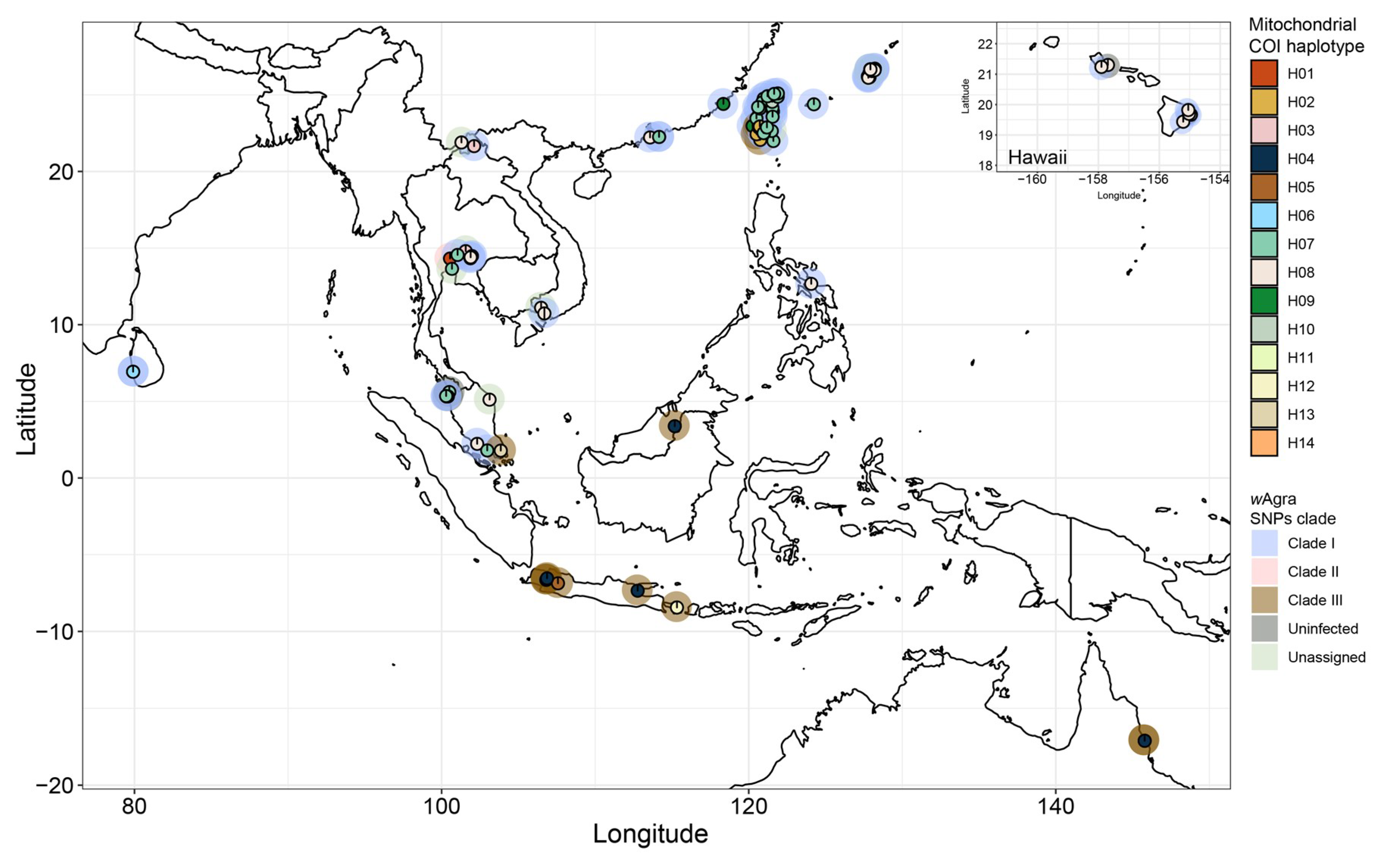
3.1. Prevalance and Strain Identity of Wolbachia
3.2. Wolbachia Genome and SNP Discovery
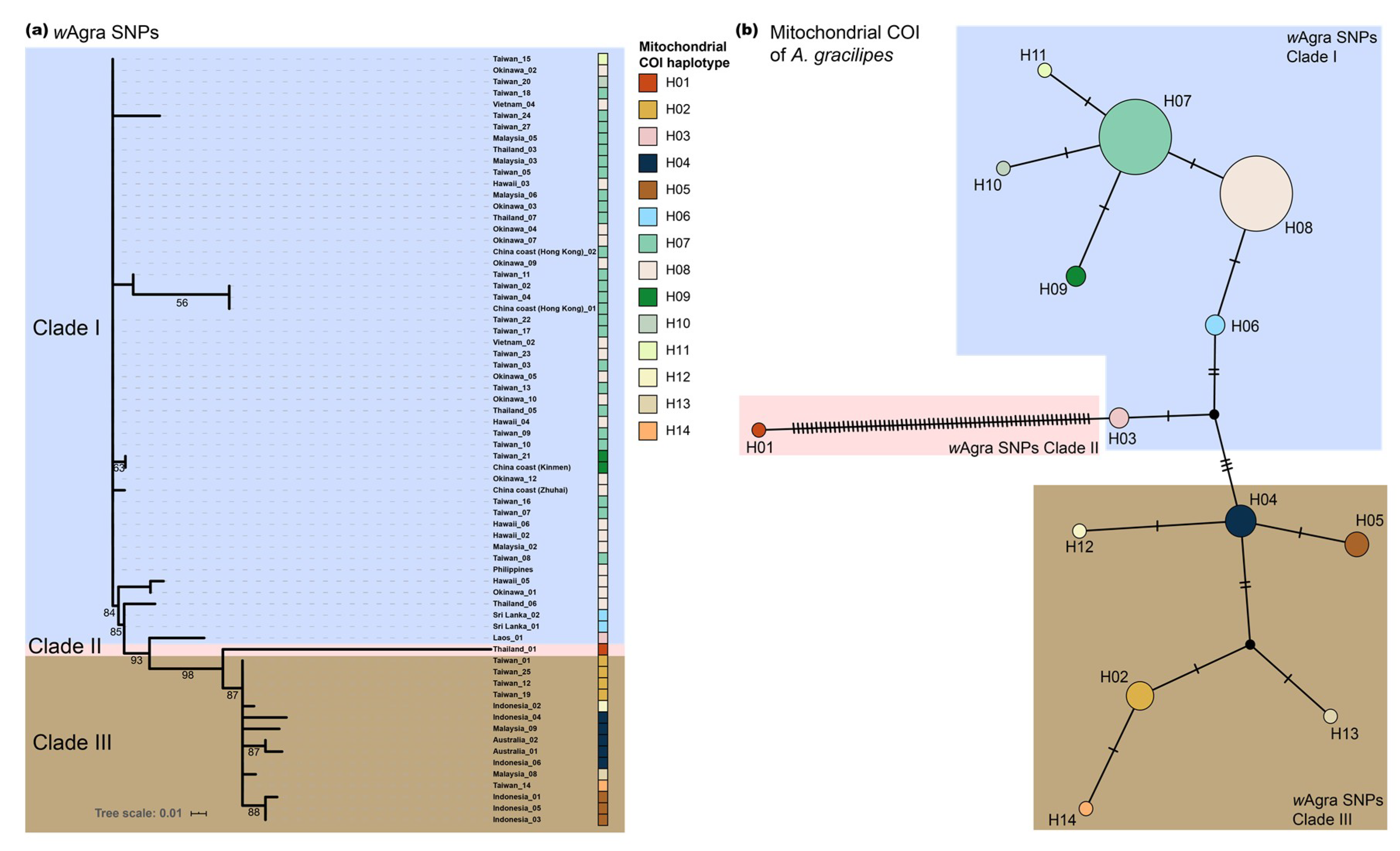
3.3. Phylogenomic Tree and Mitochondrial Network
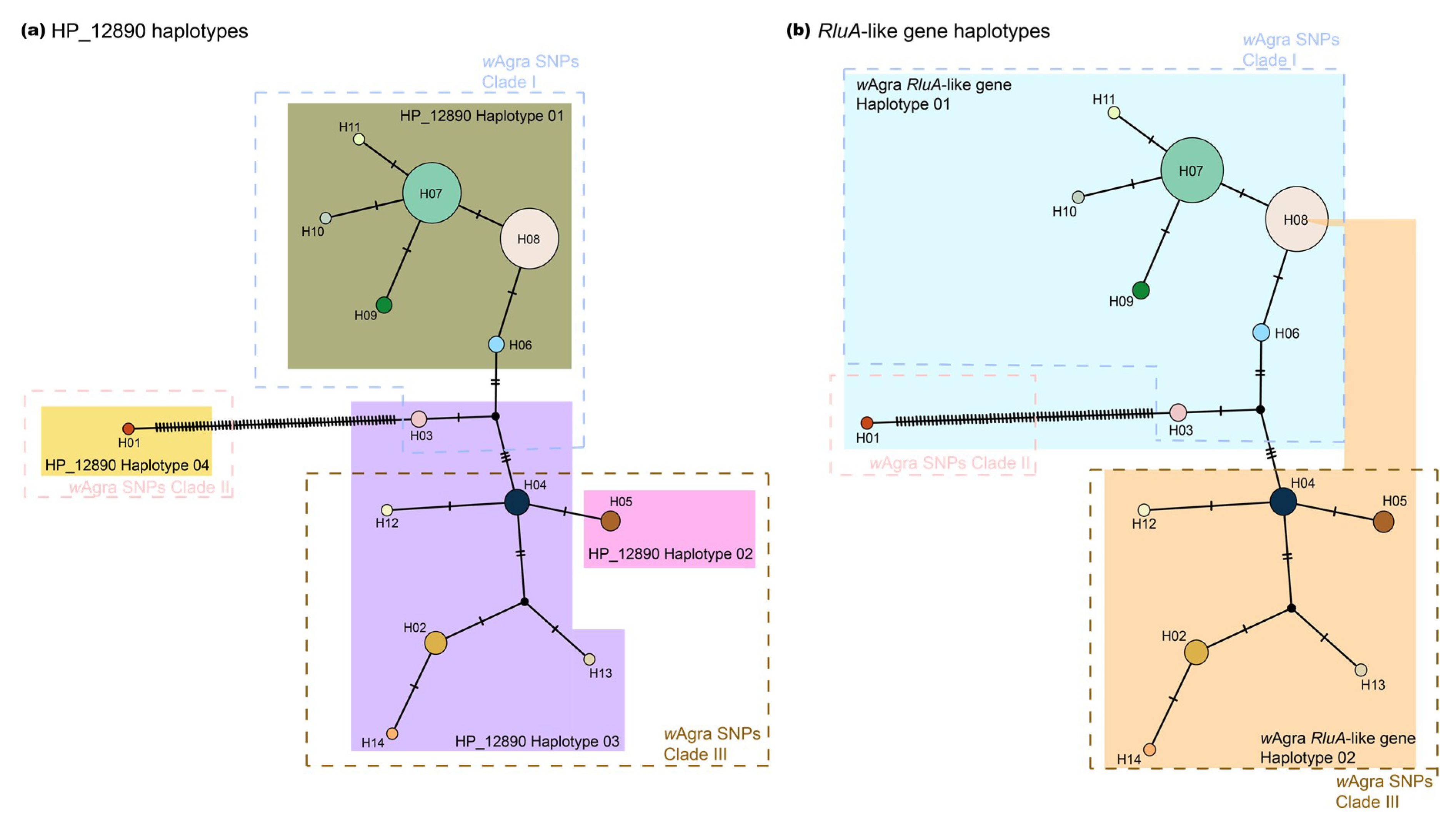
3.4. Gene Expression and Non-Synonymous ORFs
3.5. Adaptative Selections
4. Discussion
4.1. Origin of Wolbachia in Yellow Crazy Ant
4.2. High Infection Rate with a Single Wolbachia Strain
4.3. Evidence for Wolbachia–Host Coevolution
4.4. Rapid Evolution of a Hypothetical Wolbachia Gene
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Werren, J.H.; Baldo, L.; Clark, M.E. Wolbachia: Master manipulators of invertebrate biology. Nat. Rev. Microbiol. 2008, 6, 741–751. [Google Scholar] [CrossRef]
- Zug, R.; Hammerstein, P. Bad guys turned nice? A critical assessment of Wolbachia mutualisms in arthropod hosts. Biol. Rev. 2015, 90, 89–111. [Google Scholar] [CrossRef]
- Landmann, F.; Foster, J.M.; Michalski, M.L.; Slatko, B.E.; Sullivan, W. Co-evolution between an endosymbiont and its nematode host: Wolbachia asymmetric posterior localization and AP polarity establishment. PLoS Negl. Trop. Dis. 2014, 8, e3096. [Google Scholar] [CrossRef]
- Lefoulon, E.; Bain, O.; Makepeace, B.L.; d’Haese, C.; Uni, S.; Martin, C.; Gavotte, L. Breakdown of coevolution between symbiotic bacteria Wolbachia and their filarial hosts. PeerJ 2016, 4, e1840. [Google Scholar] [CrossRef]
- Balvín, O.; Roth, S.; Talbot, B.; Reinhardt, K. Co-speciation in bedbug Wolbachia parallel the pattern in nematode hosts. Sci. Rep. 2018, 8, 8797. [Google Scholar] [CrossRef]
- Newton, I.L.; Rice, D.W. The Jekyll and Hyde symbiont: Could Wolbachia be a nutritional mutualist? J. Bacteriol. 2020, 202, e00589-19. [Google Scholar] [CrossRef]
- Taylor, M.J.; Bandi, C.; Hoerauf, A. Wolbachia. Bacterial endosymbionts of filarial nematodes. Adv. Parasit. 2005, 60, 245–284. [Google Scholar]
- Riegler, M.; Stauffer, C. Wolbachia infections and superinfections in cytoplasmically incompatible populations of the European cherry fruit fly Rhagoletis cerasi (Diptera, Tephritidae). Mol. Ecol. 2002, 11, 2425–2434. [Google Scholar] [CrossRef]
- Schuler, H.; Köppler, K.; Daxböck-Horvath, S.; Rasool, B.; Krumböck, S.; Schwarz, D.; Hoffmeister, T.S.; Schlick-Steiner, B.C.; Steiner, F.M.; Telschow, A.; et al. The hitchhiker’s guide to Europe: The infection dynamics of an ongoing Wolbachia invasion and mitochondrial selective sweep in Rhagoletis cerasi. Mol. Ecol. 2016, 25, 1595–1609. [Google Scholar] [CrossRef]
- Tagami, Y.; Miura, K. Distribution and prevalence of Wolbachia in Japanese populations of Lepidoptera. Insect Mol. Ecol. 2004, 13, 359–364. [Google Scholar] [CrossRef]
- Ahmed, M.Z.; Araujo-Jnr, E.V.; Welch, J.J.; Kawahara, A.Y. Wolbachia in butterflies and moths: Geographic structure in infection frequency. Front. Zool. 2015, 12, 16. [Google Scholar] [CrossRef]
- Pontieri, L.; Schmidt, A.M.; Singh, R.; Pedersen, J.S.; Linksvayer, T.A. Artificial selection on ant female caste ratio uncovers a link between female-biased sex ratios and infection by Wolbachia endosymbionts. J. Evol. Biol. 2017, 30, 225–234. [Google Scholar] [CrossRef]
- Ün, Ç.; Schultner, E.; Manzano-Marín, A.; Flórez, L.V.; Seifert, B.; Heinze, J.; Oettler, J. Cytoplasmic incompatibility between Old and New World populations of a tramp ant. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wetterer, J.K. Worldwide distribution and potential spread of the long-legged ant, Anoplolepis gracilipes (Hymenoptera: Formicidae). Sociobiology 2005, 45, 77–97. [Google Scholar]
- Ito, F.; Asfiya, W.; Kojima, J.I. Discovery of independent-founding solitary queens in the yellow crazy ant Anoplolepis gracilipes in East Java, Indonesia (Hymenoptera: Formicidae). Entomol. Sci. 2016, 19, 312–314. [Google Scholar] [CrossRef]
- Drescher, J.; Blüthgen, N.; Schmitt, T.; Bühler, J.; Feldhaar, H. Societies drifting apart? Behavioural, genetic and chemical differentiation between supercolonies in the yellow crazy ant Anoplolepis gracilipes. PLoS ONE 2010, 5, e13581. [Google Scholar] [CrossRef]
- Gruber, M.A.; Hoffmann, B.D.; Ritchie, P.A.; Lester, P.J. Recent behavioural and population genetic divergence of an invasive ant in a novel environment. Divers. Distrib. 2012, 18, 323–333. [Google Scholar] [CrossRef]
- Tseng, S.P.; Hsu, P.W.; Lee, C.C.; Wetterer, J.K.; Hugel, S.; Wu, L.H.; Lee, C.Y.; Yoshimura, T.; Yang, C.C.S. Evidence for common horizontal transmission of Wolbachia among ants and ant crickets: Kleptoparasitism added to the list. Microorganisms 2020, 8, 805. [Google Scholar] [CrossRef]
- Sebastien, A.; Gruber, M.A.M.; Lester, P.J. Prevalence and genetic diversity of three bacterial endosymbionts (Wolbachia, Arsenophonus, and Rhizobiales) associated with the invasive yellow crazy ant (Anoplolepis gracilipes). Insectes Soc. 2012, 59, 33–40. [Google Scholar] [CrossRef]
- Baldo, L.; Hotopp, J.C.D.; Jolley, K.A.; Bordenstein, S.R.; Biber, S.A.; Choudhury, R.R.; Hayashi, C.; Maiden, M.C.J.; Tettelin, H.; Werren, J.H. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 2006, 72, 7098–7110. [Google Scholar] [CrossRef]
- PubMLST. Available online: https://pubmlst.org/wolbachia/info/protocols.shtml (accessed on 26 August 2020).
- Shoemaker, D.D.; Ahrens, M.; Sheill, L.; Mescher, M.; Keller, L.; Ross, K.G. Distribution and prevalence of Wolbachia infections in native populations of fire ant Solenopsis invicta (Hymenoptera: Formicidae). Environ. Entomol. 2003, 32, 1329–1336. [Google Scholar] [CrossRef][Green Version]
- Lee, C.C.; Wang, J.; Matsuura, K.; Yang, C.C.S. The complete mitochondrial genome of yellow crazy ant, Anoplolepis gracilipes (Hymenoptera: Formicidae). Mitochondrial DNA B Resour. 2018, 3, 622–623. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef]
- Bleidorn, C.; Gerth, M. A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol. Ecol. 2018, 94, fix163. [Google Scholar] [CrossRef]
- Kaur, R.; Siozios, S.; Miller, W.J.; Rota-Stabelli, O. Insertion sequence polymorphism and genomic rearrangements uncover hidden Wolbachia diversity in Drosophila suzukii and D. subpulchrella. Sci. Rep. 2017, 7, 14815. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Beichman, A.C.; Huerta-Sanchez, E.; Lohmueller, K.E. Using genomic data to infer historic population dynamics of nonmodel organisms. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 433–456. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 August 2020).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colón, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Dhaygude, K.; Nair, A.; Johansson, H.; Wurm, Y.; Sundström, L. The first draft genomes of the ant Formica exsecta, and its Wolbachia endosymbiont reveal extensive gene transfer from endosymbiont to host. BMC Genom. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Yang, X.; Bennett, G.M. Evolution of host support for two ancient bacterial symbionts with differentially degraded genomes in a leafhopper host. Proc. Natl. Acad. Sci. USA 2018, 11, E11691–E11700. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Goldman-Huertas, B.; Moreau, C.S.; Baldo, L.; Stahlhut, J.K.; Werren, J.H.; Pierce, N.E. Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution 2009, 63, 624–640. [Google Scholar] [CrossRef]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Wenseleers, T.; Ito, F.; Van Borm, S.; Huybrechts, R.; Volckaert, F.; Billen, J. Widespread occurrence of the microorganism Wolbachia in ants. P. Roy. Soc. B Biol. Sci. 1998, 265, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, D.D.; Ross, K.G.; Keller, L.; Vargo, E.; Werren, J.H. Wolbachia infections in native and introduced populations of fire ants (Solenopsis spp.). Insect Mol. Biol. 2000, 9, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Wenseleers, T.; Billen, J.; Boomsma, J.J. Wolbachia in leafcutter ants: A widespread symbiont that may induce male killing or incompatible matings. J. Evol. Biol. 2001, 14, 805–814. [Google Scholar] [CrossRef]
- Tsutsui, N.D.; Kauppinen, S.N.; Oyafuso, A.F.; Grosberg, R.K. The distribution and evolutionary history of Wolbachia infection in native and introduced populations of the invasive argentine ant (Linepithema humile). Mol. Ecol. 2003, 12, 3057–3068. [Google Scholar] [CrossRef]
- Frost, C.L.; Fernández-Marín, H.; Smith, J.E.; Hughes, W.O.H. Multiple gains and losses of Wolbachia symbionts across a tribe of fungus-growing ants. Mol. Ecol. 2010, 19, 4077–4085. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Martins, C.; Silva, L.M.; Martins, V.G.; Bueno, O.C. Intracellular symbiotic bacteria of Camponotus textor, Forel (Hymenoptera, Formicidae). Curr. Microbiol. 2017, 74, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Price, S.L.; de Oliveira Ramalho, M.; Moreau, C.S. Diversity of Wolbachia associated with the giant turtle ant, Cephalotes atratus. Curr. Microbiol. 2019, 76, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.P.; Wetterer, J.K.; Suarez, A.V.; Lee, C.Y.; Yoshimura, T.; Shoemaker, D.; Yang, C.C.S. Genetic diversity and Wolbachia infection patterns in a globally distributed invasive ant. Front. Genet. 2019, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Dedeine, F.; Vavre, F.; Fleury, F.; Loppin, B.; Hochberg, M.E.; Boulétreau, M. Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp. Proc. Natl. Acad. Sci. USA 2001, 98, 6247–6252. [Google Scholar] [CrossRef]
- Vorburger, C.; Gouskov, A. Only helpful when required: A longevity cost of harbouring defensive symbionts. J. Evol. Biol. 2011, 24, 1611–1617. [Google Scholar] [CrossRef]
- Oliver, K.M.; Campos, J.; Moran, N.A.; Hunter, M.S. Population dynamics of defensive symbionts in aphids. P. Roy. Soc. B Biol. Sci. 2008, 275, 293–299. [Google Scholar] [CrossRef]
- Jaenike, J.; Brekke, T.D. Defensive endosymbionts: A cryptic trophic level in community ecology. Ecol. Lett. 2011, 14, 150–155. [Google Scholar] [CrossRef]
- Cooling, M.; Gruber, M.A.M.; Hoffmann, B.D.; Sébastien, A.; Lester, P.J. A metatranscriptomic survey of the invasive yellow crazy ant, Anoplolepis gracilipes, identifies several potential viral and bacterial pathogens and mutualists. Insectes Soc. 2017, 64, 197–207. [Google Scholar] [CrossRef]
- Lee, C.C.; Lin, C.Y.; Hsu, H.W.; Yang, C.C.S. Complete genome sequences of two novel dicistroviruses detected in the yellow crazy ant, (Anoplolepis gracilipes). Arch. Vriol. 2020, 165, 2715–2719. [Google Scholar] [CrossRef]
- Cooling, M.D.; Hoffmann, B.D.; Gruber, M.A.M.; Lester, P.J. Indirect evidence of pathogen-associated altered oocyte production in queens of the invasive yellow crazy ant, Anoplolepis gracilipes, in Arnhem Land, Australia. Bull. Entomol. Res. 2018, 108, 451–460. [Google Scholar] [CrossRef]
- Hsu, H.W.; Chiu, M.C.; Lee, C.C.; Lee, C.Y.; Yang, C.C.S. The association between virus prevalence and Intercolonial aggression levels in the yellow crazy ant, Anoplolepis gracilipes (Jerdon). Insects 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Martinez, J.; Rota-Stabelli, O.; Jiggins, F.M.; Miller, W.J. Age, tissue, genotype and virus infection regulate Wolbachia levels in Drosophila. Mol. Ecol. 2020, 29, 2063–2079. [Google Scholar] [CrossRef] [PubMed]
- Frentiu, F.D.; Zakir, T.; Walker, T.; Popovici, J.; Pyke, A.T.; van den Hurk, A.; McGraw, E.A.; O’Neill, S.L. Limited dengue virus replication in field-collected Aedes aegypti mosquitoes infected with Wolbachia. PLoS Negl. Trop. Dis. 2014, 8, e2688. [Google Scholar] [CrossRef] [PubMed]
- Jiggins, F.M.; Hurst, G.D. Rapid insect evolution by symbiont transfer. Science 2011, 332, 185–186. [Google Scholar] [CrossRef]
- Himler, A.G.; Adachi-Hagimori, T.; Bergen, J.E.; Kozuch, A.; Kelly, S.E.; Tabashnik, B.E.; Chiel, E.; Duckworth, V.E.; Dennehy, T.J.; Zchori-Fein, E.; et al. Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science 2011, 332, 254–256. [Google Scholar] [CrossRef]
- Weeks, A.R.; Turelli, M.; Harcombe, W.R.; Reynolds, K.T.; Hoffmann, A.A. From parasite to mutualist: Rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 2007, 5, e114. [Google Scholar] [CrossRef]
- Keller, L.; Liautard, C.; Reuter, M.; Brown, W.D.; Sundström, L.; Chapuisat, M. Sex ratio and Wolbachia infection in the ant Formica exsecta. Heredity 2001, 87, 227–233. [Google Scholar] [CrossRef]
- Wenseleers, T.; Sundström, L.; Billen, J. Deleterious Wolbachia in the ant Formica truncorum. P. Roy. Soc. B Biol. Sci. 2002, 269, 623–629. [Google Scholar] [CrossRef]
- Singh, R.; Linksvayer, T.A. Wolbachia-infected ant colonies have increased reproductive investment and an accelerated life cycle. J. Exp. Biol. 2020, 223, jeb220079. [Google Scholar] [CrossRef]
- Kajtoch, Ł.; Kolasa, M.; Kubisz, D.; Gutowski, J.M.; Ścibior, R.; Mazur, M.A.; Holecová, M. Using host species traits to understand the Wolbachia infection distribution across terrestrial beetles. Sci. Rep. 2019, 9, 847. [Google Scholar] [CrossRef]
- Treanor, D.; Hughes, W.O.H. Limited female dispersal predicts the incidence of Wolbachia across ants (Hymenoptera: Formicidae). J. Evol. Biol. 2019, 32, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.V.; Holway, D.A.; Case, T.J. Patterns of spread in biological invasions dominated by long-distance jump dispersal: Insights from Argentine ants. Proc. Natl. Acad. Sci. USA 2001, 98, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Janicki, J.; Narula, N.; Ziegler, M.; Guénard, B.; Economo, E.P. Visualizing and interacting with large-volume biodiversity data using client–server web-mapping applications: The design and implementation of antmaps.org. Ecol. Inform. 2016, 32, 185–193. [Google Scholar] [CrossRef]
- Cheng, D.; Chen, S.; Huang, Y.; Pierce, N.E.; Riegler, M.; Yang, F.; Zeng, L.; Lu, Y.; Liang, G.; Xu, Y. Symbiotic microbiota may reflect host adaptation by resident to invasive ant species. PLoS Pathog. 2019, 15, e1007942. [Google Scholar] [CrossRef]
- Reuter, M.; Pedersen, J.S.; Keller, L. Loss of Wolbachia infection during colonisation in the invasive Argentine ant Linepithema humile. Heredity 2005, 94, 364–369. [Google Scholar] [CrossRef]
- Yang, C.C.; Yu, Y.C.; Valles, S.M.; Oi, D.H.; Chen, Y.C.; Shoemaker, D.D.; Wu, W.J.; Shih, C.J. Loss of microbial (pathogen) infections associated with recent invasions of the red imported fire ant Solenopsis invicta. Biol. Invasions 2010, 12, 3307–3318. [Google Scholar] [CrossRef]
- Biek, R.; Drummond, A.J.; Poss, M. A virus reveals population structure and recent demographic history of its carnivore host. Science 2006, 311, 538–541. [Google Scholar] [CrossRef][Green Version]
- Biek, R.; Henderson, J.C.; Waller, L.A.; Rupprecht, C.E.; Real, L.A. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proc. Natl. Acad. Sci. USA 2007, 104, 7993–7998. [Google Scholar] [CrossRef]
- Lee, C.C.; Wang, J. Rapid expansion of a highly germline-expressed mariner element acquired by horizontal transfer in the fire ant genome. Genome Biol. Evol. 2018, 10, 3262–3278. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Geographical Region | No. of Infected Colony | No. of Uninfected Colony | Infection Rate |
|---|---|---|---|
| Okinawa | 12 | 0 | 100.00% |
| Hawaii | 5 | 1 | 83.33% |
| Taiwan | 26 | 0 | 100.00% |
| Southeast coastal China (Hong Kong and Zhuhai) and Kinmen Island, Taiwan | 4 | 0 | 100.00% |
| Laos and Southwest China (Yunnan) | 2 | 0 | 100.00% |
| Vietnam | 3 | 0 | 100.00% |
| Thailand | 7 | 0 | 100.00% |
| Sri Lanka | 2 | 0 | 100.00% |
| Philippines | 1 | 0 | 100.00% |
| Malaysia | 7 | 2 | 77.78% |
| Indonesia | 6 | 0 | 100.00% |
| Australia | 2 | 0 | 100.00% |
| Total | 77 | 3 | 96.25% |
| Strain | Host Species | Super- Group | No. of Contig | Genome Size (Mb) | GC% | BUSCO Score (%) | RefSeq Assembly Accession |
|---|---|---|---|---|---|---|---|
| wAu | Drosophila simulans | A | 1 | 1.26 | 35.2 | 84.9 | GCF_000953315.1 |
| wHa | Drosophila simulans | A | 1 | 1.29 | 35.3 | 84 | GCF_000376605.1 |
| wMel | Drosophila melanogaster | A | 1 | 1.27 | 35.2 | 84.5 | GCF_000008025.1 |
| wRi | Drosophila simulans | A | 1 | 1.45 | 35.2 | 84.5 | GCF_000022285.1 |
| wAgra | Anoplolepis gracilipes | A | 96 | 1.2 | 35.2 | 85.4 | JACRYZ000000000 (This study) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-C.; Lin, C.-Y.; Tseng, S.-P.; Matsuura, K.; Yang, C.-C.S. Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis gracilipes. Microorganisms 2020, 8, 1569. https://doi.org/10.3390/microorganisms8101569
Lee C-C, Lin C-Y, Tseng S-P, Matsuura K, Yang C-CS. Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis gracilipes. Microorganisms. 2020; 8(10):1569. https://doi.org/10.3390/microorganisms8101569
Chicago/Turabian StyleLee, Chih-Chi, Chun-Yi Lin, Shu-Ping Tseng, Kenji Matsuura, and Chin-Cheng Scotty Yang. 2020. "Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis gracilipes" Microorganisms 8, no. 10: 1569. https://doi.org/10.3390/microorganisms8101569
APA StyleLee, C.-C., Lin, C.-Y., Tseng, S.-P., Matsuura, K., & Yang, C.-C. S. (2020). Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis gracilipes. Microorganisms, 8(10), 1569. https://doi.org/10.3390/microorganisms8101569