Current Status and Potential Applications of Underexplored Prokaryotes

 ,
, 
 ,
, 

Abstract
1. Introduction
2. Underexplored Prokaryotes
2.1. Definition of Underexplored Prokaryotes
2.2. Reasons for Analysing Underexplored Prokaryotes
2.3. Why Are Great Proportions of Prokaryotes Unculturable?
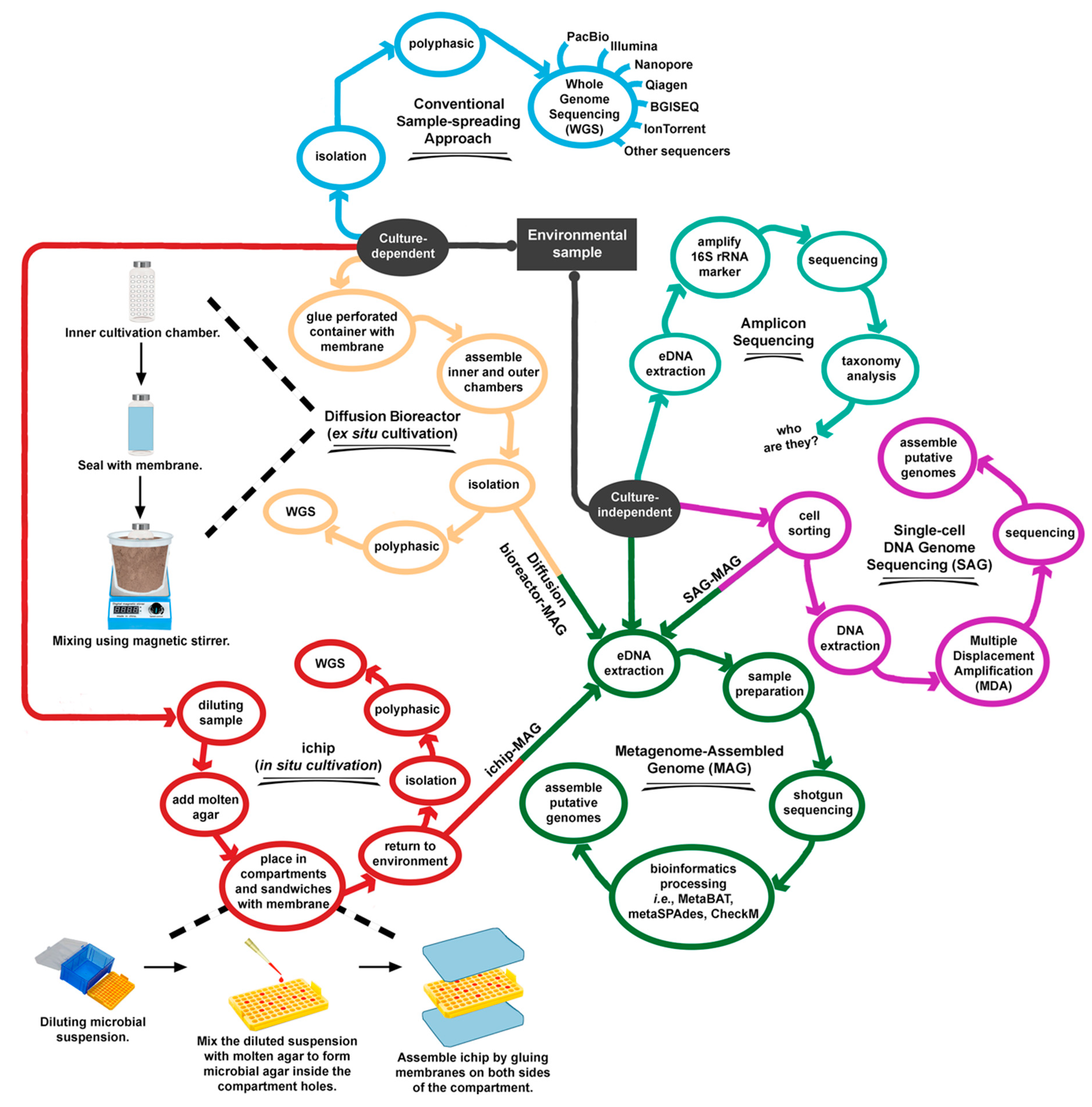
2.4. How Should Underexplored Prokaryotes Be Cultured?
2.5. Exploring Unculturable Prokaryotes Using Metagenome-Assembled Genomes (MAG)
3. Potential Applications of Underexplored Prokaryotes
3.1. Potential Applications of Culturable Rare Prokaryotes
3.2. Potential Research Topics in Unculturable Prokaryotes
4. Limitations and Future Directions of Prokaryote Discovery
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, S.; Chen, Y. Phylogenomic analysis demonstrates a pattern of rare and long-lasting concerted evolution in prokaryotes. Commun. Biol. 2018, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.T.; Tindall, B.J.; Editors, G.M.G. International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2019, 69, S1–S111. [Google Scholar]
- Trujillo, M.E.; Oren, A.; Garrity, G.M. Preparation of the Validation Lists and the role of the List Editors. Int. J. Syst. Evol. Microbiol. 2019, 69, 3–4. [Google Scholar] [CrossRef]
- Reimer, L.C.; Vetcininova, A.; Carbasse, J.S.; Söhngen, C.; Gleim, D.; Ebeling, C.; Overmann, J. BacDive in 2019: Bacterial phenotypic data for high-throughput biodiversity analysis. Nucleic Acids Res. 2019, 47, D631–D636. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and all-species Living Tree Project (LTP) taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stamatis, D.; Bertsch, J.; Ovchinnikova, G.; Katta, H.Y.; Mojica, A.; Chen, I.-M.A.; Kyrpides, N.C.; Reddy, T. Genomes OnLine database (GOLD) v.7: Updates and new features. Nucleic Acids Res. 2019, 47, D649–D659. [Google Scholar] [CrossRef]
- Field, D.; Garrity, G.; Gray, T.; Morrison, N.; Selengut, J.; Sterk, P.; Tatusova, T.; Thomson, N.; Allen, M.J.; Angiuoli, S.V.; et al. The minimum information about a genome sequence (MIGS) specification. Nat. Biotechnol. 2008, 26, 541–547. [Google Scholar] [CrossRef]
- Loman, N.J.; Pallen, M.J. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 2015, 13, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Urbieta, M.S.; Donati, E.R.; Chan, K.-G.; Shahar, S.; Sin, L.L.; Goh, K.M. Thermophiles in the genomic era: Biodiversity, science, and applications. Biotechnol. Adv. 2015, 33, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Houghton, K.M.; Carere, C.R.; Stott, M.B.; McDonald, I.R. Thermophilic methanotrophs: In hot pursuit. FEMS Microbiol. Ecol. 2019, 95, fiz125. [Google Scholar] [CrossRef]
- Lusk, B.G. Thermophiles; or, the modern prometheus: The importance of extreme microorganisms for understanding and applying extracellular electron transfer. Front. Microbiol. 2019, 10, 818. [Google Scholar] [CrossRef]
- Karp, P.D.; Ivanova, N.; Krummenacker, M.; Kyrpides, N.; Latendresse, M.; Midford, P.; Ong, W.K.; Paley, S.; Seshadri, R. A comparison of microbial genome web portals. Front. Microbiol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Alneberg, J.; Karlsson, C.M.G.; Divne, A.M.; Bergin, C.; Homa, F.; Lindh, M.V.; Hugerth, L.W.; Ettema, T.J.G.; Bertilsson, S.; Andersson, A.F.; et al. Genomes from uncultivated prokaryotes: A comparison of metagenome-assembled and single-amplified genomes. Microbiome 2018, 6, 173. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Mangot, J.F.; Logares, R.; Sánchez, P.; Latorre, F.; Seeleuthner, Y.; Mondy, S.; Sieracki, M.E.; Jaillon, O.; Wincker, P.; De Vargas, C.; et al. Accessing the genomic information of unculturable oceanic picoeukaryotes by combining multiple single cells. Sci. Rep. 2017, 7, 41498. [Google Scholar] [CrossRef]
- Kogawa, M.; Hosokawa, M.; Nishikawa, Y.; Mori, K.; Takeyama, H. Obtaining high-quality draft genomes from uncultured microbes by cleaning and co-assembly of single-cell amplified genomes. Sci. Rep. 2018, 8, 2059. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.B.; Blainey, P.C.; Schulz, F.; Woyke, T.; Horowitz, M.A.; Quake, S.R. Microfluidic-based mini-metagenomics enables discovery of novel microbial lineages from complex environmental samples. eLife 2017, 6, e26580. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, B.A.; Brian, F.; Schulz, F.; Blainey, P.C.; Woyke, T.; Quake, S.R. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc. Natl. Acad. Sci. USA 2019, 116, 5037–5044. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Strzelecka, P.M.; Ranzoni, A.M.; Cvejic, A. Dissecting human disease with single-cell omics: Application in model systems and in the clinic. Dis. Model. Mech. 2018, 11, dmm036525. [Google Scholar] [CrossRef]
- Zeng, Z.; Miao, N.; Sun, T. Revealing cellular and molecular complexity of the central nervous system using single cell sequencing. Stem Cell Res. Ther. 2018, 9, 234. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Tahon, G.; Tytgat, B.; Lebbe, L.; Carlier, A.; Willems, A. Abditibacterium utsteinense sp. nov., the first cultivated member of candidate phylum FBP, isolated from ice-free Antarctic soil samples. Syst. Appl. Microbiol. 2018, 41, 279–290. [Google Scholar] [CrossRef]
- Waite, D.W.; Vanwonterghem, I.; Rinke, C.; Parks, D.H.; Zhang, Y.; Takai, K.; Sievert, S.M.; Simon, J.; Campbell, B.J.; Hanson, T.E.; et al. Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 2017, 8, 682. [Google Scholar] [CrossRef]
- Pande, S.; Kost, C. Bacterial unculturability and the formation of intercellular metabolic networks. Trends Microbiol. 2017, 25, 349–361. [Google Scholar] [CrossRef]
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [PubMed]
- Anantharaman, K.; Brown, C.T.; Hug, L.A.; Sharon, I.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Singh, A.; Wilkins, M.J.; Karaoz, U.; et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 2016, 7, 13219. [Google Scholar] [CrossRef] [PubMed]
- Castelle, C.J.; Banfield, J.F. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 2018, 172, 1181–1197. [Google Scholar] [CrossRef]
- Youssef, N.H.; Farag, I.F.; Hahn, C.R.; Jarett, J.; Becraft, E.; Eloe-Fadrosh, E.; Lightfoot, J.; Bourgeois, A.; Cole, T.; Ferrante, S.; et al. Genomic characterization of candidate division LCP-89 reveals an atypical cell wall structure, microcompartment production, and dual respiratory and fermentative capacities. Appl. Environ. Microbiol. 2019, 85, e00110–e00119. [Google Scholar] [CrossRef]
- De Anda, V.; Zapata-Peñasco, I.; Blaz, J.; Poot-Hernández, A.C.; Contreras-Moreira, B.; González-Laffitte, M.; Gámez-Tamariz, N.; Hernández-Rosales, M.; Eguiarte, L.E.; Souza, V. Understanding the mechanisms behind the response to environmental perturbation in microbial mats: A metagenomic-network based approach. Front. Microbiol. 2018, 9, 2606. [Google Scholar] [CrossRef]
- Arrigo, K.R. Marine microorganisms and global nutrient cycles. Nature 2005, 437, 349–355. [Google Scholar] [CrossRef]
- Hu, P.; Tom, L.; Singh, A.; Thomas, B.C.; Baker, B.J.; Piceno, Y.M.; Andersen, G.L.; Banfield, J.F. Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. MBio 2016, 7. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]
- Power, J.F.; Carere, C.R.; Lee, C.K.; Wakerley, G.L.J.; Evans, D.W.; Button, M.; White, D.; Climo, M.D.; Hinze, A.M.; Morgan, X.C.; et al. Microbial biogeography of 925 geothermal springs in New Zealand. Nat. Commun. 2018, 9, 2876. [Google Scholar] [CrossRef]
- Chan, C.S.; Chan, K.-G.; Ee, R.; Hong, K.-W.; Urbieta, M.S.; Donati, E.R.; Shamsir, M.S.; Goh, K.M. Effects of physiochemical factors on prokaryotic biodiversity in Malaysian circumneutral hot springs. Front. Microbiol. 2017, 8, 1252. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Ke, T.; Li, L.; Cai, S.; Zhou, Y.; Liu, Y.; Guo, L.; Chen, L.; Zhang, D. Impacts of environmental factors on the whole microbial communities in the rhizosphere of a metal-tolerant plant: Elsholtzia haichowensis Sun. Environ. Pollut. 2018, 237, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Drivers of microbial community structure in forest soils. Appl. Microbiol. Biotechnol. 2018, 102, 4331–4338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, G.; Jiang, H.; Yang, J.; She, W.; Khan, I.; Li, W. Abundant and rare microbial biospheres respond differently to environmental and spatial factors in tibetan hot springs. Front. Microbiol. 2018, 9, 2096. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Shiller, J.; Mann, R.; Smith, G.; Yen, A.; Rodoni, B. Novel ‘Candidatus Liberibacter’ species identified in the Australian eggplant psyllid, Acizzia solanicola. Microb. Biotechnol. 2017, 10, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mckenna, B.Y.M. Antibiotics hit the orchard. Nature 2019, 567, 302–303. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Tyson, J.; Elizabeth Sockett, R. Nature knows best: Employing whole microbial strategies to tackle antibiotic resistant pathogens. Environ. Microbiol. Rep. 2017, 9, 47–49. [Google Scholar] [CrossRef]
- Tyson, J.; Sockett, R.E. Predatory bacteria: Moving from curiosity towards curative. Trends Microbiol. 2017, 25, 90–91. [Google Scholar] [CrossRef]
- Baker, M.; Negus, D.; Raghunathan, D.; Radford, P.; Moore, C.; Clark, G.; Diggle, M.; Tyson, J.; Twycross, J.; Sockett, R.E. Measuring and modelling the response of Klebsiella pneumoniae KPC prey to Bdellovibrio bacteriovorus predation, in human serum and defined buffer. Sci. Rep. 2017, 7, 8329. [Google Scholar] [CrossRef]
- Kim, W.C.; Mauborgne, R. Blue ocean strategy. In Harvard Business School Publishing Corporation; Harvard Business Review: Boston, MA, USA, 2004. [Google Scholar]
- Castelle, C.J.; Brown, C.T.; Anantharaman, K.; Probst, A.J.; Huang, R.H.; Banfield, J.F. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 2018, 16, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Towards functional characterization of archaeal genomic dark matter. Biochem. Soc. Trans. 2019, 47, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, N.; Lee, J.H.; Williams, T.A.; Offre, P.; Spang, A. Genomic diversity, lifestyles and evolutionary origins of DPANN archaea. FEMS Microbiol. Lett. 2019, 366, fnz008. [Google Scholar] [CrossRef] [PubMed]
- Raina, J.B.; Eme, L.; Pollock, F.J.; Spang, A.; Archibald, J.M.; Williams, T.A. Symbiosis in the microbial world: From ecology to genome evolution. Biol. Open 2018, 7, bio032524. [Google Scholar] [CrossRef]
- Tanaka, T.; Kawasaki, K.; Daimon, S.; Kitagawa, W.; Yamamoto, K.; Tamaki, H.; Tanaka, M.; Nakatsu, C.H.; Kamagata, Y. A hidden pitfall in the preparation of agar media undermines microorganism cultivability. Appl. Environ. Microbiol. 2014, 80, 7659–7666. [Google Scholar] [CrossRef]
- Aanderud, Z.T.; Jones, S.E.; Fierer, N.; Lennon, J.T. Resuscitation of the rare biosphere contributes to pulses of ecosystem activity. Front. Microbiol. 2015, 6, 24. [Google Scholar] [CrossRef]
- Tighe, S.; Afshinnekoo, E.; Rock, T.M.; McGrath, K.; Alexander, N.; McIntyre, A.; Ahsanuddin, S.; Bezdan, D.; Green, S.J.; Joye, S.; et al. Genomic methods and microbiological technologies for profiling novel and extreme environments for the extreme microbiome project (XMP). J. Biomol. Tech. JBT 2017, 28, 31–39. [Google Scholar] [CrossRef]
- Merino, N.; Aronson, H.S.; Bojanova, D.; Feyhl-buska, J.; Michael, L.; Zhang, S.; Giovannelli, D. Living at the extremes: Extremophiles and the limits of life in a planetary context. Front. Microbiol. 2019, 10, 780. [Google Scholar] [CrossRef]
- Lodhi, A.F.; Zhang, Y.; Adil, M.; Deng, Y. Antibiotic discovery: Combining isolation chip (ichip) technology and co-culture technique. Appl. Microbiol. Biotechnol. 2018, 102, 7333–7341. [Google Scholar] [CrossRef]
- Belanger, A.; Lewis, K.; Epstein, S.S.; Nichols, D.; Kanigan, T.; Pham, L.; Mehta, A.; Trakhtenberg, E.M.; Cahoon, N. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar]
- Chaudhary, D.K.; Khulan, A.; Kim, J. Development of a novel cultivation technique for uncultured soil bacteria. Sci. Rep. 2019, 9, 6666. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Y.; Pan, J.; Wang, F.; Li, M. Perspectives on cultivation strategies of Archaea. Microb. Ecol. 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yuan, C.G.; Xiao, M.; Tian, X.P.; Khan, I.U.; Kim, C.J.; Zhi, X.Y.; Li, W.J. Abyssicoccus albus gen. nov., sp. nov., a novel member of the family Staphylococcaceae isolated from marine sediment of the Indian Ocean. Antonie Van Leeuwenhoek 2016, 109, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Pukall, R.; Schumann, P.; Schütte, C.; Gols, R.; Dicke, M. Acaricomes phytoseiuli gen. nov., sp. nov., isolated from the predatory mite Phytoseiulus persimilis. Int. J. Syst. Evol. Microbiol. 2006, 56, 465–469. [Google Scholar] [CrossRef]
- Chen, S.; Dong, X. Acetanaero bacterium elongatum gen. nov., sp. nov., from paper mill waste water. Int. J. Syst. Evol. Microbiol. 2004, 54, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, N.; Desmarchelier, C.; Blaut, M.; Daniel, H.; Haller, D.; Clavel, T. Acetatifactor muris gen. nov., sp. nov., a novel bacterium isolated from the intestine of an obese mouse. Arch. Microbiol. 2012, 194, 901–907. [Google Scholar] [CrossRef]
- Johnson, D.B.; Stallwood, B.; Kimura, S.; Hallberg, K.B. Isolation and characterization of Acidicaldus organivorus, gen. nov., sp. nov.: A novel sulfur-oxidizing, ferric iron-reducing thermo-acidophilic heterotrophic Proteo bacterium. Arch. Microbiol. 2006, 185, 212–221. [Google Scholar] [CrossRef]
- Itoh, T.; Yamanoi, K.; Kudo, T.; Ohkuma, M.; Takashina, T. Aciditerrimonas ferrireducens gen. nov., sp. nov., an iron-reducing thermoacidophilic actinobacterium isolated from a solfataric field. Int. J. Syst. Evol. Microbiol. 2011, 61, 1281–1285. [Google Scholar] [CrossRef]
- Yamada, T.; Imachi, H.; Ohashi, A.; Harada, H.; Hanada, S.; Kamagata, Y.; Sekiguchi, Y. Bellilinea caldifistulae gen. nov., sp. nov and Longilinea arvoryzae gen. nov., sp. nov., strictly anaerobic, filamentous bacteria of the phylum Chloroflexi isolated from methanogenic propionate-degrading consortia. Int. J. Syst. Evol. Microbiol. 2007, 57, 2299–2306. [Google Scholar] [CrossRef]
- Tsuruoka, N.; Tsujimoto, Y.; Ishihara, D.; Kimura, N.; Nishino, T.; Saito, R.; Sahara, T.; Furuya, H.; Watanabe, K.; Shigeri, Y. Caenibacillus caldisaponilyticus gen. nov., sp. nov., a thermophilic, spore-forming and phospholipid-degrading bacterium isolated from acidulocompost. Int. J. Syst. Evol. Microbiol. 2016, 66, 2684–2690. [Google Scholar]
- Wagner, I.D.; Ahmed, S.; Zhao, W.; Zhang, C.L.; Romanek, C.S.; Rohde, M.; Wiegel, J. Caldanaerovirga acetigignens gen. nov., sp. nov., an anaerobic xylanolytic, alkalithermophilic bacterium isolated from Trego Hot Spring, Nevada, USA. Int. J. Syst. Evol. Microbiol. 2009, 59, 2685–2691. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sorokin, D.Y.; Rakitin, A.L.; Gumerov, V.M.; Beletsky, A.V.; Sinninghe Damsté, J.S.; Mardanov, A.V.; Ravin, N.V. Phenotypic and genomic properties of Chitinispirillum alkaliphilum gen. nov., sp. nov., a haloalkaliphilic anaerobic chitinolytic bacterium representing a novel class in the phylum Fibrobacteres. Front. Microbiol. 2016, 7, 407. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, P.; Hoffmann, B. Dichotomicrobium thermohalophilum, gen. nov., spec, nov., budding prosthecate bacteria from the solar lake (Sinai) and some related strains. Syst. Appl. Microbiol. 1989, 149, 547–556. [Google Scholar] [CrossRef]
- Labeda, D.P.; Kroppenstedt, R.M.; Euzéby, J.P.; Tindall, B.J. Proposal of Goodfellowiella gen. nov. to replace the illegitimate genus name Goodfellowia labeda and Kroppenstedt 2006. Int. J. Syst. Evol. Microbiol. 2008, 58, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Jung, J.Y.; Chae, H.B.; Park, W.; Jeon, C.O. Hwanghaeicola aestuarii gen. nov., sp. nov., a moderately halophilic bacterium isolated from a tidal flat of the Yellow Sea. Int. J. Syst. Evol. Microbiol. 2010, 60, 2877–2881. [Google Scholar] [CrossRef][Green Version]
- Zhang, C.F.; Ai, M.J.; Wang, J.X.; Liu, S.W.; Zhao, L.L.; Su, J.; Sun, C.H.; Yu, L.Y.; Zhang, Y.Q. Herbihabitans rhizosphaerae gen. nov., sp. nov., a member of the family Pseudonocardiaceae isolated from rhizosphere soil of the herb Limonium sinense (Girard). Int. J. Syst. Evol. Microbiol. 2016, 66, 4156–4161. [Google Scholar]
- Rekha, P.D.; Young, C.C.; Kämpfer, P.; Martin, K.; Arun, A.B.; Chen, W.M.; Lai, W.A.; Chao, J.H.; Shen, F.T. Jhaorihella thermophila gen. nov., sp. nov., a moderately thermophilic bacterium isolated from a coastal hot spring. Int. J. Syst. Evol. Microbiol. 2011, 61, 1544–1548. [Google Scholar] [CrossRef]
- Jumas-Bilak, E.; Carlier, J.P.; Jean-Pierre, H.; Citron, D.; Bernard, K.; Damay, A.; Gay, B.; Teyssier, C.; Campos, J.; Marchandin, H. Jonquetella anthropi gen. nov., sp. nov., the first member of the candidate phylum “Synergistetes” isolated from man. Int. J. Syst. Evol. Microbiol. 2007, 57, 2743–2748. [Google Scholar] [CrossRef]
- Ara, I.; Kudo, T. Krasilnikovia gen. nov., a new member of the family Micromonosporaceae and description of Krasilnikovia cinnamonea sp. nov. Actinomycetologica 2007, 21, 1–10. [Google Scholar] [CrossRef]
- Weon, H.Y.; Kim, B.Y.; Kwon, S.W.; Park, I.C.; Cha, I.B.; Tindall, B.J.; Stackebrandt, E.; Trüper, H.G.; Go, S.J. Leadbetterella byssophila gen. nov., sp. nov., isolated from cotton-waste composts for the cultivation of oyster mushroom. Int. J. Syst. Evol. Microbiol. 2005, 55, 2297–2302. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cao, Y.; Zhou, G.; Yin, J.; Qiu, J. Mangrovicoccus ximenensis gen. nov., sp. nov., isolated from mangrove forest sediment. Int. J. Syst. Evol. Microbiol. 2018, 68, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Vorobev, A.V.; Baani, M.; Doronina, N.V.; Brady, A.L.; Liesack, W.; Dunfield, P.F.; Dedysh, S.N. Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int. J. Syst. Evol. Microbiol. 2011, 61, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, T.N.; Dallimore, J.; Lünsdorf, H.; Heipieper, H.J.; Golyshin, P.N. Monaibacterium marinum, gen. nov, sp. nov, a new member of the Alphaproteo bacteria isolated from seawater of Menai Straits, Wales, UK. Int. J. Syst. Evol. Microbiol. 2017, 67, 3310–3317. [Google Scholar] [PubMed]
- Zhang, L.; Shen, X.; Liu, Y.; Li, S. Nafulsella turpanensis gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from soil. Int. J. Syst. Evol. Microbiol. 2013, 63, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Asker, D.; Beppu, T.; Ueda, K. Nubsella zeaxanthinifaciens gen. nov., sp. nov., a zeaxanthin-producing bacterium of the family Sphingobacteriaceae isolated from freshwater. Int. J. Syst. Evol. Microbiol. 2008, 58, 601–606. [Google Scholar] [CrossRef]
- Podosokorskaya, O.A.; Bonch-Osmolovskaya, E.A.; Novikov, A.A.; Kolganova, T.V.; Kublanov, I.V. Ornatilinea apprima gen. nov., sp. nov., a cellulolytic representative of the class Anaerolineae. Int. J. Syst. Evol. Microbiol. 2013, 63, 86–92. [Google Scholar] [CrossRef]
- Bengelsdorf, F.R.; Poehlein, A.; Schiel-Bengelsdorf, B.; Daniel, R.; Dürre, P. Genome sequence of the acetogenic bacterium Oxobacter pfennigii DSM 3222T. Genome Announc. 2015, 3, e01408–e01415. [Google Scholar] [CrossRef]
- Xia, X.; Wu, S.; Han, Y.; Liao, S.; Wang, G. Pelobium manganitolerans gen. nov., sp. nov., isolated from sludge of a manganese mine. Int. J. Syst. Evol. Microbiol. 2016, 66, 4954–4959. [Google Scholar]
- Kulichevskaya, I.S.; Ivanova, A.O.; Belova, S.E.; Baulina, O.I.; Bodelier, P.L.E.; Rijpstra, W.I.C.; Sinninghe Damsté, J.S.; Zavarzin, G.A.; Dedysh, S.N. Schlesneria paludicola gen. nov., sp. nov., the first acidophilic member of the order Planctomycetales, from Sphagnum-dominated boreal wetlands. Int. J. Syst. Evol. Microbiol. 2007, 57, 2680–2687. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Rossello-Mora, R.; Amann, R. Moving the cataloguing of the “uncultivated majority” forward. Syst. Appl. Microbiol. 2019, 42, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Danczak, R.E.; Johnston, M.D.; Kenah, C.; Slattery, M.; Wrighton, K.C.; Wilkins, M.J. Members of the Candidate Phyla Radiation are functionally differentiated by carbon- and nitrogen-cycling capabilities. Microbiome 2017, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Z.; Goh, K.M.; Evans, P.N.; Liu, L.; Mao, Y.; Hugenholtz, P.; Tyson, G.W.; Li, W.; Zhang, T. Further expansion of methane metabolism in the Archaea. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kadnikov, V.V.; Mardanov, A.V.; Beletsky, A.V.; Rakitin, A.L.; Frank, Y.A.; Karnachuk, O.V.; Ravin, N.V. Phylogeny and physiology of candidate phylum BRC1 inferred from the first complete metagenome-assembled genome obtained from deep subsurface aquifer. Syst. Appl. Microbiol. 2019, 42, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Wilkins, L.G.E.; Ettinger, C.L.; Jospin, G.; Eisen, J.A. Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal pools in Kamchatka, Russia. Sci. Rep. 2019, 9, 3059. [Google Scholar] [CrossRef]
- Dong, X.; Greening, C.; Rattray, J.E.; Chakraborty, A.; Chuvochina, M.; Mayumi, D.; Dolfing, J.; Li, C.; Brooks, J.M.; Bernard, B.B.; et al. Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 2019, 10, 1816. [Google Scholar] [CrossRef]
- Kasanke, C.P.; Collins, R.E.; Leigh, M.B. Identification and characterization of a dominant sulfolane-degrading Rhodoferax sp. via stable isotope probing combined with metagenomics. Sci. Rep. 2019, 9, 3121. [Google Scholar] [CrossRef]
- Sharp, C.; Kleiner, M.; Gordon, P.M.K.; Pon, R.T.; Dong, X. A shared core microbiome in soda lakes separated by large distances. Nat. Commun. 2019, 10, 4230. [Google Scholar]
- Wang, Y.; Niu, Q.; Zhang, X.; Liu, L.; Wang, Y.; Chen, Y.; Negi, M.; Figeys, D.; Li, Y.-Y.; Zhang, T. Exploring the effects of operational mode and microbial interactions on bacterial community assembly in a one-stage partial-nitritation anammox reactor using integrated multi-omics. Microbiome 2019, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Liu, J.; Fang, Y.; Hedlund, B.P.; Lian, Z.H.; Huang, L.Y.; Li, J.T.; Huang, L.N.; Li, W.J.; Jiang, H.C.; et al. Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 2019, 13, 2044–2057. [Google Scholar] [CrossRef] [PubMed]
- Tsementzi, D.; Rodriguez-R, L.M.; Ruiz-Perez, C.A.; Meziti, A.; Hatt, J.K.; Konstantinidis, K.T. Ecogenomic characterization of widespread, closely-related SAR11 clades of the freshwater genus “Candidatus Fonsibacter” and proposal of Ca. Fonsibacter lacus sp. nov. Syst. Appl. Microbiol. 2019, 42, 495–505. [Google Scholar] [CrossRef]
- Castelle, C.J.; Brown, C.T.; Thomas, B.C.; Williams, K.H.; Banfield, J.F. Unusual respiratory capacity and nitrogen metabolism in a Parcubacterium (OD1) of the Candidate Phyla Radiation. Sci. Rep. 2017, 7, 40101. [Google Scholar] [CrossRef] [PubMed]
- Kadnikov, V.V.; Mardanov, A.V.; Beletsky, A.V.; Karnachuk, O.V.; Ravin, N.V. Genome of the candidate phylum Aminicenantes bacterium from a deep subsurface thermal aquifer revealed its fermentative saccharolytic lifestyle. Extremophiles 2019, 23, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; White, R.A.; Visscher, P.T.; Charlesworth, J.C.; Vázquez-Campos, X.; Burns, B.P. Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes. ISME J. 2018, 12, 2619–2639. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, J.; Espinoza, S.; Viver, T.; Alcamán-Arias, M.E.; Trefault, N.; Rosselló-Móra, R.; Díez, B. Temperature modulates Fischerella thermalis ecotypes in Porcelana Hot Spring. Syst. Appl. Microbiol. 2018, 41, 531–543. [Google Scholar] [CrossRef]
- Fortney, N.W.; He, S.; Converse, B.J.; Boyd, E.S.; Roden, E.E. Investigating the composition and metabolic potential of microbial communities in chocolate pots hot springs. Front. Microbiol. 2018, 9, 2075. [Google Scholar] [CrossRef]
- Press, M.O.; Roehe, R.; Snelling, T.J.; Dewhurst, R.J.; Watson, M.; Stewart, R.D.; Auffret, M.D.; Warr, A.; Langford, K.W.; Liachko, I.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar]
- Hugerth, L.W.; Larsson, J.; Alneberg, J.; Lindh, M.V.; Legrand, C.; Pinhassi, J.; Andersson, A.F. Metagenome-assembled genomes uncover a global brackish microbiome. Genome Biol. 2015, 16, 279. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dasgupta, S.; Guo, Z.; Chen, S.; Xu, H.; Ta, K.; Bai, S.; Du, M.; Peng, X.; Chen, M. Microplastics contaminate the deepest part of the world’s ocean. Geochemical Perspect. Lett. 2018, 9, 1–5. [Google Scholar]
- Wei, R.; Zimmermann, W. Microbial enzymes for the recycling of recalcitrant petroleum-based plastics: How far are we? Microb. Biotechnol. 2017, 10, 1308–1322. [Google Scholar] [CrossRef] [PubMed]
- Oberbeckmann, S.; Labrenz, M. Marine microbial assemblages on microplastics: Diversity, adaptation, and role in degradation. Ann. Rev. Mar. Sci. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 2016, 351, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Skaf, M.S.; Austin, H.P.; Crowley, M.F.; McGeehan, J.E.; Silveira, R.L.; Donohoe, B.S.; Pollard, B.C.; Michener, W.E.; Kearns, F.L.; Duman, R.; et al. Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc. Natl. Acad. Sci. USA 2018, 115, E4350–E4357. [Google Scholar]
- Joo, S.; Seo, H.; Cho, I.J.; Lee, S.Y.; Choi, S.Y.; Son, H.F.; Shin, T.J.; Sagong, H.-Y.; Kim, K.-J. Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation. Nat. Commun. 2018, 9, 382. [Google Scholar] [CrossRef]
- Wei, R.; Föllner, C.; Then, J.; Zimmermann, W.; Sträter, N.; Roth, C.; Oeser, T. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl. Microbiol. Biotechnol. 2014, 98, 7815–7823. [Google Scholar]
- Zhang, Y.; Wang, L.; Chen, J.; Wu, J. Enhanced activity toward PET by site-directed mutagenesis of Thermobifida fusca cutinase-CBM fusion protein. Carbohydr. Polym. 2013, 97, 124–129. [Google Scholar] [CrossRef]
- Danso, D.; Schmeisser, C.; Chow, J.; Zimmermann, W.; Wei, R.; Leggewie, C.; Li, X.; Hazen, T.; Streita, W.R. New insights into the function and gobal distribution of polyethylene terephthalate (PET)-degrading cacteria and enzymes in marine and terrestrial metagenomes. Appl. Environ. Microbiol. 2018, 53, 1689–1699. [Google Scholar]
- Baltz, R.H. Gifted microbes for genome mining and natural product discovery. J. Ind. Microbiol. Biotechnol. 2017, 44, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Idris, H.; Goodfellow, M.; Sanderson, R.; Asenjo, J.A.; Bull, A.T. Actinobacterial rare biospheres and dark matter revealed in habitats of the Chilean Atacama desert. Sci. Rep. 2017, 7, 8373. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, S.; Willems, A.; Tahon, G. Uncovering the uncultivated majority in Antarctic soils: Toward a synergistic approach. Front. Microbiol. 2019, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Cavicchioli, R.; Charlton, T.; Ertan, H.; Omar, S.M.; Siddiqui, K.S.; Williams, T.J. Biotechnological uses of enzymes from psychrophiles. Microb. Biotechnol. 2011, 4, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Pikuta, E.V.; Menes, R.J.; Bruce, A.M.; Lyu, Z.; Patel, N.B.; Liu, Y.; Hoover, R.B.; Busse, H.J.; Lawson, P.A.; Whitman, W.B. Raineyella antarctica gen. nov., sp. nov., a psychrotolerant, D-amino-acid-utilizing anaerobe isolated from two geographic locations of the Southern Hemisphere. Int. J. Syst. Evol. Microbiol. 2016, 66, 5529–5536. [Google Scholar] [PubMed]
- Sheridan, P.P.; Loveland-Curtze, J.; Miteva, V.I.; Brenchley, J.E. Rhodoglobus vestalii gen. nov., sp. nov., a novel psychrophilic organism isolated from an Antarctic Dry Valley Lake. Int. J. Syst. Evol. Microbiol. 2003, 53, 985–994. [Google Scholar] [CrossRef]
- Li, H.R.; Yu, Y.; Luo, W.; Zeng, Y.X. Marisediminicola antarctica gen. nov., sp. nov., an Actinobacterium isolated from the Antarctic. Int. J. Syst. Evol. Microbiol. 2010, 60, 2535–2539. [Google Scholar] [CrossRef]
- Podosokorskaya, O.A.; Kadnikov, V.V.; Gavrilov, S.N.; Mardanov, A.V.; Merkel, A.Y.; Karnachuk, O.V.; Ravin, N.V.; Bonch-Osmolovskaya, E.A.; Kublanov, I.V. Characterization of Melioribacter roseus gen. nov., sp. nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae. Environ. Microbiol. 2013, 15, 1759–1771. [Google Scholar] [CrossRef]
- Rakitin, A.L.; Ermakova, A.Y.; Ravin, N.V. Novel endoxylanases of the moderately thermophilic polysaccharide-degrading bacterium Melioribacter roseus. J. Microbiol. Biotechnol. 2015, 25, 1476–1484. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Bonch-Osmolovskaya, E.A.; Gavrilov, S.N.; Ravin, N.V.; Podosokorskaya, O.A.; Kadnikov, V.V.; Beletsky, A.V.; Kublanov, I.V. Genomic analysis of Melioribacter roseus, facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi group. PLoS ONE 2013, 8, e53047. [Google Scholar]
- Hameed, A.; Shahina, M.; Lin, S.Y.; Sridhar, K.R.; Young, L.S.; Lee, M.R.; Chen, W.M.; Chou, J.H.; Young, C.C. Siansivirga zeaxanthinifaciens gen. nov., sp. nov., a novel zeaxanthin-producing member of the family Flavobacteriaceae isolated from coastal seawater of Taiwan. FEMS Microbiol. Lett. 2012, 333, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Shahina, M.; Huang, H.C.; Lai, W.A.; Lin, S.Y.; Stothard, P.; Young, C.C. Complete genome sequence of Siansivirga zeaxanthinifaciens CC-SAMT-1T, a flavobacterium isolated from coastal surface seawater. Mar. Genomics 2018, 37, 21–25. [Google Scholar] [CrossRef]
- Liao, Z.; Holtzapple, M.; Yan, Y.; Wang, H.; Li, J.; Zhao, B. Insights into xylan degradation and haloalkaline adaptation through whole-genome analysis of Alkalitalea saponilacus, an anaerobic haloalkaliphilic bacterium capable of secreting novel halostable xylanase. Genes 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, S. Alkalitalea saponilacus gen. nov., sp. nov., an obligately anaerobic, alkaliphilic, xylanolytic bacterium from a meromictic soda lake. Int. J. Syst. Evol. Microbiol. 2012, 62, 2618–2623. [Google Scholar] [CrossRef] [PubMed]
- Yaakop, A.S.; Chan, K.-G.; Ee, R.; Lim, Y.L.; Lee, S.-K.; Manan, F.A.; Goh, K.M. Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 2016, 6, 33660. [Google Scholar] [CrossRef]
- Liew, K.J.; Lim, L.; Woo, H.Y.; Chan, K.G.; Shamsir, M.S.; Goh, K.M. Purification and characterization of a novel GH1 beta-glucosidase from Jeotgalibacillus malaysiensis. Int. J. Biol. Macromol. 2018, 115, 1094–1102. [Google Scholar] [CrossRef]
- Gomes, J.; Gomes, I.; Terler, K.; Gubala, N.; Ditzelmüller, G.; Steiner, W. Optimisation of culture medium and conditions for α-L-arabinofuranosidase production by the extreme thermophilic eubacterium Rhodothermus marinus. Enzyme Microb. Technol. 2000, 27, 414–422. [Google Scholar] [CrossRef]
- Hachem, M.A.; Karlsson, E.N.; Bartonek-Roxâ, E.; Raghothama, S.; Simpson, P.J.; Gilbert, H.J.; Williamson, M.P.; Holst, O. Carbohydrate-binding modules from a thermostable Rhodothermus marinus xylanase: Cloning, expression and binding studies. Biochem. J. 2000, 345, 53–60. [Google Scholar] [CrossRef]
- Park, M.-J.; Oh, J.H.; Yang, S.-H.; Kwon, K.K. Roseithermus sacchariphilus gen. nov., sp. nov. and proposal of Salisaetaceae fam. nov., representing new family in the order Rhodothermales. Int. J. Syst. Evol. Microbiol. 2019, 69, 1213–1219. [Google Scholar] [CrossRef]
- Liew, K.-J.; Teo, S.C.; Shamsir, M.S.; Sani, R.K.; Chong, C.S.; Chan, K.G.; Goh, K.M. Complete genome sequence of Rhodothermaceae bacterium RA with cellulolytic and xylanolytic activities. 3 Biotech 2018, 8, 376. [Google Scholar] [CrossRef]
- Goh, K.M.; Chan, K.-G.; Lim, S.W.; Liew, K.J.; Chan, C.S.; Shamsir, M.S.; Ee, R.; Adrian, T.-G.-S. Genome analysis of a new Rhodothermaceae strain isolated from a hot spring. Front. Microbiol. 2016, 7, 1109. [Google Scholar] [CrossRef] [PubMed]
- Jun, K.; Yi, C.; Shahir, M.; Kumar, R.; Shiong, C.; Mau, K. Heterologous expression, purification and biochemical characterization of a new endo-1, 4-β-xylanase from Rhodothermaceae RA bacterium RA. Protein Expr. Purif. 2019, 164, 105464. [Google Scholar]
- Teo, S.C.; Liew, K.J.; Shamsir, M.S.; Chong, C.S.; Bruce, N.C.; Chan, K.-G.; Goh, K.M. Characterizing a halo-tolerant GH10 xylanase from Roseithermus sacchariphilus strain RA and its CBM-truncated variant. Int. J. Mol. Sci. 2019, 20, E2284. [Google Scholar] [CrossRef] [PubMed]
- Izumori, K. Izumoring: A strategy for bioproduction of all hexoses. J. Biotechnol. 2006, 124, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Raychaudhuri, U.; Chakraborty, R. Artificial sweeteners—A review. J. Food Sci. Technol. 2014, 51, 611–621. [Google Scholar] [CrossRef]
- Wasaki, J.; Taguchi, H.; Senoura, T.; Akasaka, H.; Watanabe, J.; Kawaguchi, K.; Komata, Y.; Hanashiro, K.; Ito, S. Identification and distribution of cellobiose 2-epimerase genes by a PCR-based metagenomic approach. Appl. Microbiol. Biotechnol. 2015, 99, 4287–4295. [Google Scholar] [CrossRef]
- Pimentel, A.C.; Ematsu, G.C.G.; Liberato, M.V.; Paixão, D.A.A.; Franco Cairo, J.P.L.; Mandelli, F.; Tramontina, R.; Gandin, C.A.; de Oliveira Neto, M.; Squina, F.M.; et al. Biochemical and biophysical properties of a metagenome-derived GH5 endoglucanase displaying an unconventional domain architecture. Int. J. Biol. Macromol. 2017, 99, 384–393. [Google Scholar] [CrossRef]
- Kotik, M.; Vanacek, P.; Kunka, A.; Prokop, Z.; Damborsky, J. Metagenome-derived haloalkane dehalogenases with novel catalytic properties. Appl. Microbiol. Biotechnol. 2017, 101, 6385–6397. [Google Scholar] [CrossRef]
- Rong, Z.; Cui, H.-L.; Huo, Y.-Y.; Xu, X.-W.; Jian, S.-L.; Cheng, H. Two novel deep-sea sediment metagenome-derived esterases: Residue 199 is the determinant of substrate specificity and preference. Microb. Cell Factories 2018, 17, 16. [Google Scholar]
- Angelov, A.; Pham, V.T.T.; Übelacker, M.; Brady, S.; Leis, B.; Pill, N.; Brolle, J.; Mechelke, M.; Moerch, M.; Henrissat, B.; et al. A metagenome-derived thermostable β-glucanase with an unusual module architecture which defines the new glycoside hydrolase family GH148. Sci. Rep. 2017, 7, 17306. [Google Scholar] [CrossRef]
- Tao, L.Y.; Gong, J.S.; Su, C.; Jiang, M.; Li, H.; Li, H.; Lu, Z.M.; Xu, Z.H.; Shi, J.S. Mining and expression of a metagenome-derived keratinase responsible for biosynthesis of silver nanoparticles. ACS Biomater. Sci. Eng. 2018, 4, 1307–1315. [Google Scholar] [CrossRef]
- Silva-Portela, R.C.B.; Carvalho, F.M.; Pereira, C.P.M.; De Souza-Pinto, N.C.; Modesti, M.; Fuchs, R.P.; Agnez-Lima, L.F. ExoMeg1: A new exonuclease from metagenomic library. Sci. Rep. 2016, 6, 19712. [Google Scholar] [CrossRef] [PubMed]
- Toyama, D.; de Morais, M.A.B.; Ramos, F.C.; Zanphorlin, L.M.; Tonoli, C.C.C.; Balula, A.F.; de Miranda, F.P.; Almeida, V.M.; Marana, S.R.; Ruller, R.; et al. A novel β-glucosidase isolated from the microbial metagenome of Lake Poraquê (Amazon, Brazil). Biochim. Biophys. Acta—Proteins Proteom. 2018, 1866, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Berini, F.; Casciello, C.; Marcone, G.L.; Marinelli, F. Metagenomics: Novel enzymes from non-culturable microbes. FEMS Microbiol. Lett. 2017, 364, fnx211. [Google Scholar] [CrossRef]
- Tiwari, R.; Nain, L.; Labrou, N.E.; Shukla, P. Bioprospecting of functional cellulases from metagenome for second generation biofuel production: A review. Crit. Rev. Microbiol. 2018, 44, 244–257. [Google Scholar] [CrossRef]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell. Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef]
- Hedlund, B.P.; Dodsworth, J.A.; Murugapiran, S.K.; Rinke, C.; Woyke, T. Impact of single-cell genomics and metagenomics on the emerging view of extremophile “microbial dark matter”. Extremophiles 2014, 18, 865–875. [Google Scholar] [CrossRef]
- Mende, D.R.; Aylward, F.O.; Eppley, J.M.; Nielsen, T.N.; DeLong, E.F. Improved environmental genomes via integration of metagenomic and single-cell assemblies. Front. Microbiol. 2016, 7, 143. [Google Scholar] [CrossRef]
- Berdy, B.; Spoering, A.L.; Ling, L.L.; Epstein, S.S. In situ cultivation of previously uncultivable microorganisms using the ichip. Nat. Protoc. 2017, 12, 2232–2242. [Google Scholar] [CrossRef]
- Han, K.; Li, Z.F.; Peng, R.; Zhu, L.P.; Zhou, T.; Wang, L.G.; Li, S.G.; Zhang, X.B.; Hu, W.; Wu, Z.H.; et al. Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci. Rep. 2013, 3, 2101. [Google Scholar] [CrossRef]
- Riley, A.B.; Kim, D.; Hansen, A.K. Genome sequence of “Candidatus Carsonella ruddii” strain BC, a nutritional endosymbiont of Bactericera cockerelli. Genome Announc. 2017, 5, e00236-17. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [PubMed]
- Goh, K.M.; Gan, H.M.; Chan, K.-G.; Chan, G.F.; Shahar, S.; Chong, C.S.; Kahar, U.M.; Chai, K.P. Analysis of Anoxybacillus genomes from the aspects of lifestyle adaptations, prophage diversity, and carbohydrate metabolism. PLoS ONE 2014, 9, e90549. [Google Scholar] [CrossRef] [PubMed]
- Saw, J.H.; Mountain, B.W.; Feng, L.; Omelchenko, M.V.; Hou, S.; Saito, J.A.; Stott, M.B.; Li, D.; Zhao, G.; Wu, J.; et al. Encapsulated in silica: Genome, proteome and physiology of the thermophilic bacterium Anoxybacillus flavithermus WK1. Genome Biol. 2008, 9, R161. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, N.; Selvaraj, A.; Kumaar, L.K.; Kumar, G.R. Genome wide re-annotation of Caldicellulosiruptor saccharolyticus with new insights into genes involved in biomass degradation and hydrogen production. PLoS ONE 2015, 10, e0133183. [Google Scholar] [CrossRef] [PubMed]
- Maeder, D.L.; Weiss, R.B.; Dunn, D.M.; Cherry, J.L.; González, J.M.; DiRuggiero, J.; Robb, F.T. Divergence of the hyperthermophilic archaea Pyrococcus furiosus and P. horikoshii inferred from complete genomic sequences. Genetics 1999, 152, 1299–1305. [Google Scholar] [PubMed]
- Da Costa, W.L.O.; De Aragão Araújo, C.L.; Dias, L.M.; De Sousa Pereira, L.C.; Alves, J.T.C.; Araújo, F.A.; Folador, E.L.; Henriques, I.; Silva, A.; Folador, A.R.C. Functional annotation of hypothetical proteins from the Exiguobacterium antarcticum strain B7 reveals proteins involved in adaptation to extreme environments, including high arsenic resistance. PLoS ONE 2018, 13, e0198965. [Google Scholar] [CrossRef] [PubMed]
- Ijaq, J.; Malik, G.; Kumar, A.; Das, P.S.; Meena, N.; Bethi, N.; Sundararajan, V.S.; Suravajhala, P. A model to predict the function of hypothetical proteins through a nine-point classification scoring schema. BMC Bioinform. 2019, 20, 14. [Google Scholar] [CrossRef]
- Naveed, M.; Tehreem, S.; Usman, M.; Chaudhry, Z.; Abbas, G. Structural and functional annotation of hypothetical proteins of human adenovirus: Prioritizing the novel drug targets. BMC Res. Notes 2017, 10, 706. [Google Scholar] [CrossRef]
- Kim, D.W.; Lee, K.S.; Chi, Y.M. Crystal structure of hypothetical protein PA4202 from Pseudomonas aeruginosa PAO1 in complex with nitroethane as a nitroalkane substrate. Biochem. Biophys. Res. Commun. 2018, 503, 330–337. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
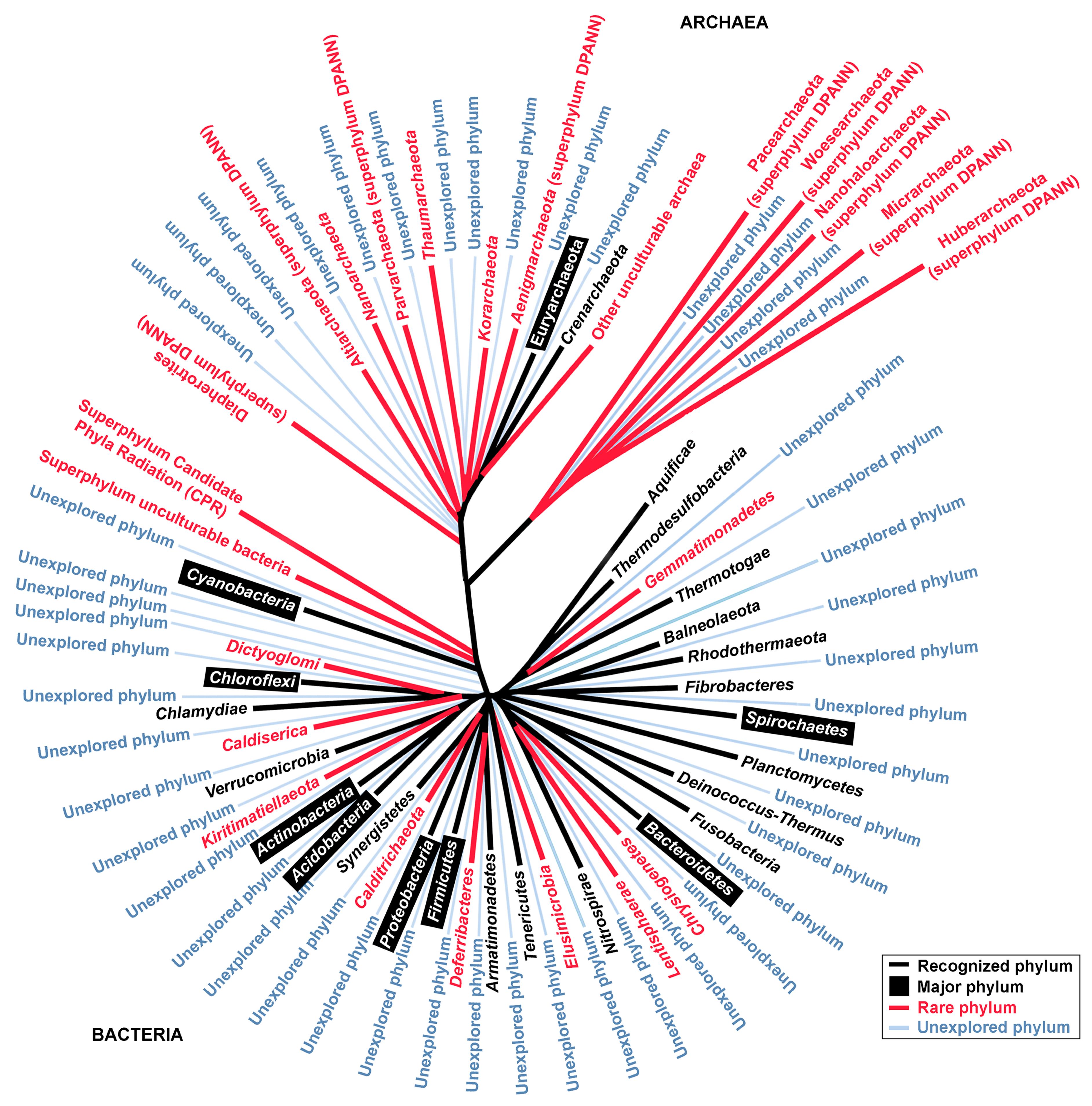
| Phyla and Candidate Phyla | No. of 16S rRNA Sequences | Remarks | |
|---|---|---|---|
| NCBI Taxonomy Database | Silva Database | ||
| Domain bacteria | |||
| Recognized bacterial phylum | |||
| Acidobacteria | 162,567 | 14,534 | Major phylum |
| Actinobacteria | 4,719,261 | 60,510 | Major phylum |
| Aquificae | 23,441 | 346 | |
| Armatimonadetes | 16,593 | 752 | |
| Bacteroidetes | 1,116,583 | 55,663 | Major phylum |
| Balneolaeota | 1589 | n/a | |
| Caldiserica | 3630 | 95 | Underexplored culturable, 1 genus, 1 type strain |
| Calditrichaeota | 11,735 | 275 | Underexplored culturable, 1 genus, 1 type strain |
| Chlamydiae | 147,839 | 450 | |
| Chlorobi | 6655 | n/a | |
| Chloroflexi | 168,644 | 9245 | Major phylum |
| Chrysiogenetes | 1253 | 12 | Underexplored culturable, 3 genera, 4 type strains |
| Cyanobacteria | 481,641 | 13,906 | Major phylum |
| Deferribacteres | 1968 | 134 | |
| Deinococcus–Thermus | 57,234 | 948 | |
| Dictyoglomi | 296 | 11 | Underexplored culturable, 1 genus, 2 type strains |
| Elusimicrobia | 15,992 | 435 | Underexplored culturable, 2 genus, 2 type strains |
| Fibrobacteres | 8597 | 751 | |
| Firmicutes | 10,435,846 | 149,757 | Major phylum |
| Fusobacteria | 57,398 | 2216 | |
| Gemmatimonadetes | 34,287 | 21,185 | Underexplored culturable, 1 genus, 2 type strains |
| Kiritimatiellaeota | 1149 | 975 | Underexplored culturable, 1 genus, 1 type strain |
| Lentisphaerae | 8098 | 469 | Underexplored culturable, 3 genera, 5 type strain |
| Nitrospirae | 48,424 | 1297 | |
| Planctomycetes | 110,685 | 9014 | Major phylum |
| Proteobacteria | 28,570,321 | 238,949 | |
| Rhodothermaeota | 731 | n/a | |
| Spirochaetes | 373,114 | 4253 | Major phylum |
| Synergistetes | 22,879 | 1152 | |
| Tenericutes | 80,247 | 2561 | |
| Thermodesulfobacteria | 4586 | n/a | |
| Thermotogae | 22,371 | 303 | |
| Verrucomicrobia | 131,082 | 4419 | |
| Domain bacteria | |||
| Superphylum unculturable bacteria | |||
| Abditibacterium | 120 | 155 | Abditibacterium utsteinense is the first representative of candidate phylum FBP [28] |
| Abyssubacteria | 291 | n/a | Bacteria candidate |
| Acetothermia | 3411 | 165 | Bacteria candidate phylum |
| Aegiribacteria | 36 | 52 | Bacteria candidate |
| Aerophobetes | 4393 | 66 | Bacteria candidate phylum |
| Atribacteria | 9087 | 578 | Bacteria candidate phylum |
| Aureabacteria | 106 | n/a | Bacteria candidate |
| Calescamantes | 1004 | 12 | Bacteria candidate |
| Cloacimonetes | 10,048 | 301 | Bacteria candidate |
| Coprothermobacteraeota | 811 | 76 | Bacteria candidate |
| Dadabacteria | 3979 | 169 | Bacteria candidate phylum |
| Dependentiae | 1756 | 580 | Candidate bacteria phylum |
| Desantisbacteria | 2580 | 2 | Bacteria candidate phylum |
| Edwardsbacteria | 133 | 4 | Bacteria candidate phylum |
| Entotheonellaeota | n/a | 168 | Bacteria candidate |
| Epsilonbacteraeota | n/a | 5422 | The class was proposed as phylum [29] |
| Fermentibacteria | 1426 | n/a | Bacteria candidate |
| Fervidibacteria | 698 | 4 | Bacteria candidate phylum |
| Firestonebacteria | 770 | 3 | Bacteria candidate |
| Halanaerobiaeota | n/a | 270 | Bacteria candidate |
| Hydrogenedentes | 1984 | 271 | Bacteria candidate |
| Hydrothermae | 1987 | 38 | Bacteria candidate phylum |
| Kryptonia | 4416 | n/a | Bacteria candidate |
| Latescribacteria | 6535 | 497 | Bacteria candidate |
| Lindowbacteria | 194 | 1 | Bacteria candidate phylum |
| Margulisbacteria | 1455 | 86 | Bacteria candidate |
| Marinimicrobia | 13,480 | 554 | Bacteria candidate |
| Modulibacteria | n/a | 255 | Bacteria candidate phylum |
| Nitrospinae | 24,196 | 2167 | Bacteria candidate phylum |
| Omnitrophicaeota | 15,814 | 507 | Bacteria candidate |
| Patescribacteria | n/a | 4521 | Bacteria candidate phylum |
| Poribacteria | 3664 | 49 | Bacteria candidate phylum |
| Rokubacteria | 33,383 | 380 | Bacteria candidate phylum |
| Schekmanbacteria | 1628 | 40 | Bacteria candidate phylum |
| Tectomicrobia | 17,004 | n/a | Bacteria candidate phylum |
| Thermosulfidibacteraeota | n/a | 3 | Bacteria candidate phylum |
| Zixibacteria | 7432 | 203 | Bacteria candidate |
| Unassigned | 6,575,288 | 54 | |
| Superphylum Candidate Phyla Radiation (CPR) | 952 | n/a | |
| Domain archaea | |||
| Recognized archaeal phylum | |||
| Crenarchaeota | 67,854 | 4611 | |
| Euryarchaeota | 307,348 | 12,957 | Major phylum |
| Korarchaeota | 5406 | 55 | Underexplored unculturable |
| Nanoarchaeota | 1689 | 869 | Underexplored unculturable |
| Thaumarchaeota | 39,720 | 4809 | Underexplored culturable, 1 genus, 1 type strain |
| Domain archaea | |||
| Candidate archaea | |||
| Aenigmarchaeota | 3324 | 42 | Underexplored unculturable DPANN |
| Altiarchaeota | n/a | 935 | Underexplored unculturable DPANN |
| Diapherotrites | 1034 | 1169 | Underexplored unculturable DPANN |
| Huberarchaeota | n/a | 909 | Underexplored unculturable DPANN |
| Micrarchaeota | 2844 | n/a | Underexplored unculturable DPANN |
| Nanohaloarchaeota | 3300 | n/a | Underexplored unculturable DPANN |
| Pacearchaeota | 2916 | n/a | Underexplored unculturable DPANN |
| Parvarchaeota | 371 | 201 | Underexplored unculturable DPANN |
| Woesearchaeota | 6468 | n/a | Underexplored unculturable DPANN |
| Other unculturable archaea | 33,442 | 2886 | Including TACK and Asgard group |
| No. | Phyla | Genus | Total Species a | Total Number of Related Articles b |
|---|---|---|---|---|
| 1 | Actinobacteria | Streptomyces | 848 | 35,008 |
| 2 | Firmicutes | Bacillus | 377 | 168,001 |
| 3 | Proteobacteria | Pseudomonas | 254 | 162,460 |
| 4 | Firmicutes | Paenibacillus | 240 | 1861 |
| 5 | Firmicutes | Lactobacillus | 237 | 49,320 |
| 6 | Firmicutes | Clostridium | 229 | 54,265 |
| 7 | Bacteroidetes | Flavobacterium | 208 | 5555 |
| 8 | Actinobacteria | Mycobacterium | 198 | 114,210 |
| 9 | Proteobacteria | Vibrio | 147 | 35,798 |
| 10 | Actinobacteria | Corynebacterium | 132 | 19,605 |
| 11 | Firmicutes | Streptococcus | 129 | 142,792 |
| 12 | Tenericutes | Mycoplasma | 127 | 28,075 |
| 13 | Proteobacteria | Sphingomonas | 127 | 3051 |
| 14 | Proteobacteria | Burkholderia | 122 | 11,383 |
| 15 | Actinobacteria | Nocardia | 119 | 7969 |
| 16 | Proteobacteria | Rhizobium | 112 | 24,085 |
| 17 | Bacteroidetes | Chryseobacterium | 112 | 1278 |
| 18 | Actinobacteria | Microbacterium | 110 | 1576 |
| 19 | Actinobacteria | Nocardioides | 103 | 435 |
| 20 | Proteobacteria | Halomonas | 102 | 1411 |
| No. | Species | Phyla | Patents | Paper Citations |
|---|---|---|---|---|
| 1 | Corynebacterium glutamicum ATCC 13032 | Actinobacteria | 315 | 478 |
| 2 | Staphylococcus aureus subsp. aureus Rosenbach ATCC 6538 | Firmicutes | 184 | 610 |
| 3 | Synechocystis sp. PCC 6803 | Cyanobacteria | 170 | 4615 |
| 4 | Corynebacterium glutamicum ATCC 13869 | Actinobacteria | 131 | 59 |
| 5 | Bacillus subtilis subsp. spizizenii ATCC 6633 | Firmicutes | 125 | 1292 |
| 6 | Escherichia coli ATCC 25922 | Proteobacteria | 113 | 3594 |
| 7 | Staphylococcus aureus subsp. aureus ATCC 29213 | Firmicutes | 108 | 1809 |
| 8 | Brevibacterium flavum ATCC 14067 | Actinobacteria | 103 | 59 |
| 9 | bCandida albicans ATCC 10231 | Ascomycota | 93 | 488 |
| 10 | Escherichia coli ATCC 8739 | Proteobacteria | 79 | 315 |
| Phylum/Family | Monotypic Name | Source | Growth Condition | Genome Size (Mb) | NCBI Genome Accession no. | Ref. |
|---|---|---|---|---|---|---|
| Abditibacteriota/Abitibacteriaceae | Abditibacter iumutsteinense | Antartic soil | psychrophile | 3.61 | GCA_002973605 | [28] |
| Firmicutes/Staphylococcaceae | Abyssicoccus albus | deep sea sediment | mesophilic | 1.8 | GCA_003815035 | [65] |
| Actinobacteria/Micrococcaceae | Acaricomes phytoseiuli | predatory mite | mesophile, slow grower | 2.4 | NZ_AQXM00000000 | [66] |
| Firmicutes/Ruminococcaceae | Acetanaerobacterium elongatum | wastewater | mesophile, anaerobe | 2.9 | NZ_FNID00000000 | [67] |
| Firmicutes/Clostridia | Acetatifactor muris | cecum of mouse | mesophile, anaerobe | 6.0 | NZ_OFSM00000000 | [68] |
| Proteobacteria/Acetobacteraceae | Acidicaldus organivorans | hot spring | thermophile | 2.89 | GCA_000759655 | [69] |
| Actinobacteria/Acidimicrobiaceae | Aciditerrimonas ferrireducens | solfataric soil | thermophile | 1.18 | GCA_001311945 | [70] |
| Chloroflexi/Anaerolineaceae | Bellilinea caldifistulae | thermophilic digester sludge | thermophile, anaerobe | 3.66 | NZ_LGHJ00000000 | [71] |
| Firmicutes/Sporolactobacillaceae | Caenibacillus caldisaponilyticus | Acidic compost | thermophile | 3.35 | GCA_002003465 | [72] |
| Firmicutes/Thermodesulfobiaceae | Caldanaerovirga acetigignens | hot spring | thermophile, anaerobe | 2.26 | GCA_900142995 | [73] |
| Fibrobacteres/Chitinispirillaceae | Chitinispirillum alkaliphilum | hypersaline soda lake | mesophile, anaerobic | 4.4 | GCA_001045525 | [74] |
| Proteobacteria/Hyphomicrobiaceae | Dichotomicrobium thermohalophilum | solar lake | thermophile | 2.99 | NZ_QXDF00000000 | [75] |
| Actinobacteria/Pseudonocardiaceae | Goodfellowiella coeruleoviolacea | soil | mesophile | 9.3 | GCA_000715825 | [76] |
| Proteobacteria/Rhodobacteraceae | Hwanghaeicola aestuarii | tidal sediment | mesophile | 4.54 | GCA_003253995 | [77] |
| Actinobacteria/Pseudonocardiaceae | Herbihabitans rhizosphaerae | soil | mesophile | 6.64 | GCA_004216555 | [78] |
| Proteobacteria/Rhodobacteraceae | Jhaorihella thermophila | coastal hot spring | moderate thermophile | 3.77 | GCA_900108275 | [79] |
| Synergistetes/Synergistaceae | Jonquetella anthropi | human cyst | mesophile, anaerobe | 1.68 | NZ_AGRU00000000 | [80] |
| Actinobacteria/Micromonosporaceae | Krasilnikovia cinnamomea | soil | mesophile | 7.62 | GCA_004217545 | [81] |
| Bacteroidetes/Cytophagaceae | Leadbetterella byssophila | cotton waste compost | mesophile | 4.06 | CP002305 | [82] |
| Proteobacteria/Rhodobacteraceae | Mangrovicoccus ximenensis | mangrove forest | halotolerant, mesophile | 5.97 | GCA_003056725 | [83] |
| Proteobacteria/Beijerinckiaceae | Methyloferula stellata | acidic peat soil | psychrophile | 4.24 | NZ_ARWA00000000 | [84] |
| Proteobacteria/Rhodobacteraceae | Monaibacterium marinum | sea water | mesophile | 3.73 | GCA_900231835 | [85] |
| Bacteroidetes/Flammeovirgaceae | Nafulsella turpanensis | soil | mesophile | 4.81 | GCA_000346615 | [86] |
| Bacteroidetes/Sphingobacteriaceae | Nubsella zeaxanthinifaciens | fresh water | mesophile | 4.25 | GCA_003313335 | [87] |
| Chloroflexi/Anaerolineaceae | Ornatilinea apprima | deep well | thermophile, anaerobe, | 4.35 | GCA_001306115 | [88] |
| Firmicutes/Clostridiaceae | Oxobacter pfennigii | rumen of cattle | mesophile, anaerobe | 4.51 | GCA_001317355 | [89] |
| Bacteroidetes/Sphingobacteriaceae | Pelobium manganitolerans | sludge of a mine | mesophile | 3.93 | GCA_003609575 | [90] |
| Planctomycetes/Planctomycetaceae | Schlesneria paludicola | sphagnum peat | mesophile | 8.67 | GCA_000255655 | [91] |
| Source | Major Bioinformatics Tools | Purpose/Major Findings | NCBI Bioproject Accession no., Unless Stated | Year/Reference |
|---|---|---|---|---|
| Aquifer | SPAdes, CONCOCT, CheckM | Analyze the genome of Candidate bacterial phylum BRC1. | SRR710274, CP030759 | 2019 [95] |
| Hot spring | SPAdes, Kaiju, CheckM | Expanding the understanding (diversity, phylogenetic, and functional) of microbiome in two well-studied hot springs in Kamchatka, Russia. | PRJNA419931 | 2019 [98] |
| Deep-sea | BBmap, MetaBAT, CheckM | Recovered 82 MAGs affiliated with 21 different archaeal and bacterial phyla from petroleum seepage. Authors proposed that acetate and hydrogen are the central intermediates underpinning community interactions and biogeochemical cycling. | PRJNA415828, PRJNA485648 | 2019 [99] |
| Aquifer | bbduk in the bbmap package, SPAdes, VizBin, CheckM | Assembled the genome of Rhodoferax sp. However, the authors were not able to confirm that this bacterium can degrade sulfolane in the contaminated aquifer. | 181102 (JGI IMG/ER) | 2019 [100] |
| Soda lakes | BBnorm, MetaSpades, MetaBat, CheckM | Used metagenomics and metaproteomics to provide a comprehensive molecular characterization of a phototrophic microbial mat microbiome. | PRJNA377096 | 2019 [101] |
| Lab-scale reactor | CLC de novo assembler, CheckM | Explaining the shifts in microbial community structures using 16S rRNA metagenome, MAGs, and metaproteomic data. | PRJNA471375 | 2019 [102] |
| Artificial acid mine drainage | SPAdes, CheckM, ESOM | Describe taxonomy and ecological role of a new order Ca. Acidulodesulfobacterales (Sva0485 clade). | PRJNA517999 | 2019 [103] |
| Freshwater | SolexaQA++, Scythe, IDBA-UD, MetaBAT, MASH, MiGA, CheckM | Explain poorly understood Ca. Pelagibacterales (SAR11 clade IIIb). | PRJNA495371, PRJNA214105, PRJNA497294 | 2019 [104] |
| Aquifer | IDBA-UD, ggKbase, ABAWACA, ESOM | Reconstruct the genome of Candidate Parcunitrobacter nitroensis (OD1), and Candidate phylum Aminicenantes (OP8). | LBUF00000000, QUAH00000000 | 2019, 2017 [105,106] |
| Ocean | Minimus2, BinSanity, CheckM | Reconstruct the genome of 2,631 genomes, as part of Tara Oceans project. | PRJNA391943 | 2018 [107] |
| Bay | MEGAHIT, CheckM, RAST, Phylosift, JspeciesWS | Assembled 87 MAGs including archaeal Asgard group (Thorarchaetoa and Lokiarchaeota). Reveal potential microbial interactions. | 4761314.3–4761727.3, 4762868.3–4762965.3 (MGRAST) | 2018 [108] |
| Hot spring | IDBA, MaxBin, CheckM | Reconstruct the genome of cyanobacteria Fischerella thermalis. | NA382437 | 2018 [109] |
| Hot spring | metaSPAdes, CONCOCT, SNAP, CheckM | To relate MAGs’ extracellular electron transfer systems with iron redox-based metabolisms | 3300010938 and 3300014149 (IMG/M ER) | 2018 [110] |
| Cow rumen | MetaBAT, dRep, CheckM | Improve the understanding of taxonomic structure of rumen microbiome. To mine novel carbohydrate degrading enzymes. | PRJEB21624 | 2018 [111] |
| Offshore station | Refers to original articles | Compare SAG and MAG for samples collected from the same site. | PRJEB21451, LIAK00000000–LIDO00000000 | 2018 [18,112] |
| Aquifer | MetaBAT, AMPHORA2 | Propose the carbon and nitrogen cycling functions for 71 putative CPR genomes. | SRX2896383 | 2017 [93] |
| Metadata obtained from SRA database | CLC de novo assembler, MetaBAT, CheckM, RefineM | Expand the understanding of phylogenetic and genomes of uncultivated bacteria and archaea. | PRJNA34875 | 2017 [17] |
| Oil reservoir | EMIRGE, IDBA-UD, ggKbase, ESOM | Describe the metabolic process of candidate phyla. | SRP057267, PRJNA278302 | 2016 [38] |
| Aquifer | IDBA-UD, ggKbase, ABAWACA, ESOM | Reconstruct the metabolism to understand the geochemical cycling Construct the CPR genomes and notice that the genomes are small and lacked many important biosynthesis pathways. | PRJNA273161, PRJNA288027 | 2016, 2015 [33,54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goh, K.M.; Shahar, S.; Chan, K.-G.; Chong, C.S.; Amran, S.I.; Sani, M.H.; Zakaria, I.I.; Kahar, U.M. Current Status and Potential Applications of Underexplored Prokaryotes. Microorganisms 2019, 7, 468. https://doi.org/10.3390/microorganisms7100468
Goh KM, Shahar S, Chan K-G, Chong CS, Amran SI, Sani MH, Zakaria II, Kahar UM. Current Status and Potential Applications of Underexplored Prokaryotes. Microorganisms. 2019; 7(10):468. https://doi.org/10.3390/microorganisms7100468
Chicago/Turabian StyleGoh, Kian Mau, Saleha Shahar, Kok-Gan Chan, Chun Shiong Chong, Syazwani Itri Amran, Mohd Helmi Sani, Iffah Izzati Zakaria, and Ummirul Mukminin Kahar. 2019. "Current Status and Potential Applications of Underexplored Prokaryotes" Microorganisms 7, no. 10: 468. https://doi.org/10.3390/microorganisms7100468
APA StyleGoh, K. M., Shahar, S., Chan, K.-G., Chong, C. S., Amran, S. I., Sani, M. H., Zakaria, I. I., & Kahar, U. M. (2019). Current Status and Potential Applications of Underexplored Prokaryotes. Microorganisms, 7(10), 468. https://doi.org/10.3390/microorganisms7100468