Development of Versatile Vectors for Heterologous Expression in Bacillus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction of Fragment Exchange (FX)-Compatible Vectors for Bacillus Expression
2.2. Sub-Cloning of gfp and apr Genes to the FX-Compatible Bacillus Vectors
2.3. Transformation of Bacillus subtilis by Natural Competence
2.4. Heterologous Expression of Green Fluorescent Protein (GFP) and Subtilisins in Bacillus subtilis
2.5. GFP Fluorescence Measurement
2.6. Detection of Recombinant Subtilisins by Immunoblotting
2.7. Subtilisin Activity Assays
3. Results
3.1. Preparation of FX-Compatible Bacillus Vectors
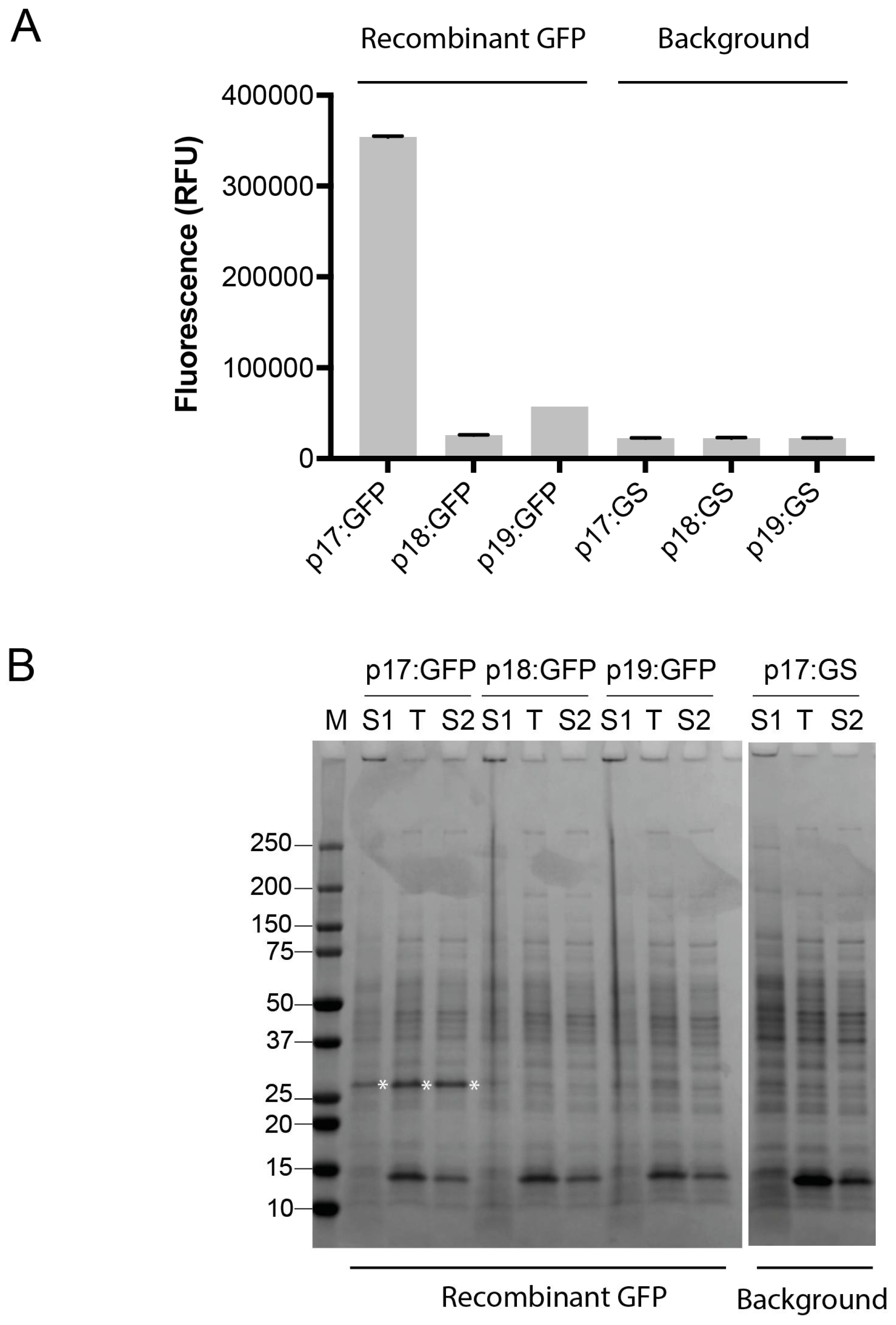
3.2. Expression of GFP in Bacillus subtilis
3.3. Validation of Vectors by Expression of Active Recombinant Subtilisin Proteins in Bacillus subtilis
3.4. Initial Characterisation of Four Different Subtilisin Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kwon, K.; Hasseman, J.; Latham, S.; Grose, C.; Do, Y.; Fleischmann, R.D.; Pieper, R.; Peterson, S.N. Recombinant expression and functional analysis of proteases from Streptococcus pneumoniae, Bacillus anthracis, and Yersinia pestis. BMC Biochem. 2011, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Sroga, G.E.; Dordick, J.S. A strategy for in vivo screening of subtilisin E reaction specificity in E. coli periplasm. Biotechnol. Bioeng. 2002, 78, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Leunissen, J.A. Subtilases: The superfamily of subtilisin-like serine proteases. Protein Sci. 1997, 6, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Güntelberg, A.V.; Ottesen, M. Preparation of Crystals containing the Plakalbumin-forming Enzyme from Bacillus subtilis. Nature 1952, 170, 802. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Families of serine peptidases. Methods Enzymol. 1994, 244, 19–61. [Google Scholar] [PubMed]
- Gupta, R.; Beg, Q.K.; Lorenz, P. Bacterial alkaline proteases: Molecular approaches and industrial applications. Appl. Microbiol. Biotechnol. 2002, 59, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Doi, R.H. Determination of the signal peptidase cleavage site in the preprosubtilisin of Bacillus subtilis. J. Biol. Chem. 1986, 261, 10176–10181. [Google Scholar] [PubMed]
- Wells, J.A.; Ferrari, E.; Henner, D.J.; Estell, D.A.; Chen, E.Y. Cloning, sequencing, and secretion of Bacillus amyloliquefaciens subtilisin in Bacillus subtilis. Nucleic Acids Res. 1983, 11, 7911–7925. [Google Scholar] [CrossRef] [PubMed]
- Vasantha, N.; Thompson, L.D.; Rhodes, C.; Banner, C.; Nagle, J.; Filpula, D. Genes for alkaline protease and neutral protease from Bacillus amyloliquefaciens contain a large open reading frame between the regions coding for signal sequence and mature protein. J. Bacteriol. 1984, 159, 811–819. [Google Scholar] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2009, 38, D211–D222. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Antelmann, H.; Jongbloed, J.D.H.; Braun, P.G.; Darmon, E.; Dorenbos, R.; Dubois, J.-Y.F.; Westers, H.; Zanen, G.; Quax, W.J.; et al. Proteomics of protein secretion by Bacillus subtilis: Separating the ‘secrets’ of the secretome. Microbiol. Mol. Biol. Rev. 2004, 68, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, H.; Takagi, H.; Inouye, M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J. Biol. Chem. 1987, 262, 7859–7864. [Google Scholar] [PubMed]
- Ohta, Y.; Hojo, H.; Aimoto, S.; Kobayashi, T.; Zhu, X.; Jordan, F.; Inouye, M. Pro-peptide as an intramolecular chaperone: Renaturation of denatured subtilisin E with a synthetic pro-peptide. Mol. Microbiol. 1991, 5, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.L.; Ohta, Y.; Jordan, F.; Inouye, M. Pro-sequence of subtilisin can guide the refolding of denatured subtilisin in an intermolecular process. Nature 1989, 339, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, H.; Inouye, M. In vitro processing of pro-subtilisin produced in Escherichia coli. J. Biol. Chem. 1988, 263, 12959–12963. [Google Scholar] [PubMed]
- Bjerga, G.E.K.; Arsın, H.; Larsen, Ø.; Puntervoll, P.; Kleivdal, H.T. A rapid solubility-optimized screening procedure for recombinant subtilisins in E. coli. J. Biotechnol. 2016, 222, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, M.; Coscolín, C.; Santiago, G.; Chow, J.; Stogios, P.J.; Bargiela, R.; Gertler, C.; Navarro-Fernández, J.; Bollinger, A.; Thies, S.; et al. The INMARE Consortium, Determinants and Prediction of Esterase Substrate Promiscuity Patterns. ACS Chem. Biol. 2018, 13, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Martínez-Martínez, M.; Bargiela, R.; Streit, W.R.; Golyshina, O.V.; Golyshin, P.N. Estimating the success of enzyme bioprospecting through metagenomics: Current status and future trends. Microb. Biotechnol. 2016, 9, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, L.P.; Sowdhamini, R. Genome-wide survey of prokaryotic serine proteases: Analysis of distribution and domain architectures of five serine protease families in prokaryotes. BMC Genom. 2008, 9, 549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Phan, T.T.P.; Schumann, W. Analysis and application of Bacillus subtilis sortases to anchor recombinant proteins on the cell wall. AMB Express. 2011, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed]
- Geertsma, E.R.; Dutzler, R. A versatile and efficient high-throughput cloning tool for structural biology. Biochemistry 2011, 50, 3272–3278. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Couturier, M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 1992, 226, 735–745. [Google Scholar] [CrossRef]
- Hartley, J.L.; Temple, G.F.; Brasch, M.A. DNA cloning using in vitro site-specific recombination. Genome Res. 2000, 10, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Busso, D.; Delagoutte-Busso, B.; Moras, D. Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 2005, 343, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Stammen, S.; Müller, B.K.; Korneli, C.; Biedendieck, R.; Gamer, M.; Franco-Lara, E.; Jahn, D. High-yield intra- and extracellular protein production using Bacillus megaterium. Appl. Environ. Microbiol. 2010, 76, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Van den Ent, F.; Löwe, J. RF cloning: A restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 2006, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.A.; Leiros, H.-K.S.; Bjerga, G.E.K. A high-throughput, restriction-free cloning and screening strategy based on ccdB-gene replacement. Microb. Cell Fact. 2014, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Bjerga, G.E.K.; Williamson, A.K. Cold shock induction of recombinant Arctic environmental genes. BMC Biotechnol. 2015, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, C.; Spizizen, J. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 1961, 81, 741–746. [Google Scholar] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Bolhuis, A.; Jongbloed, J.D.; Bron, S.; van Dijl, J.M. Signal peptide-dependent protein transport in Bacillus subtilis: A genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 2000, 64, 515–547. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.D. Recombining low homology, functionally rich regions of bacterial subtilisins by combinatorial fragment exchange. PLoS ONE 2011, 6, e24319. [Google Scholar] [CrossRef] [PubMed]
- Tindbaek, N.; Svendsen, A.; Oestergaard, P.R.; Draborg, H. Engineering a substrate-specific cold-adapted subtilisin. Protein Eng. Des. Sel. 2004, 17, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Jacobovitch, Y.; Dantes, A.; Bernheim, R.; Peleg, Y. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J. Struct. Biol. 2010, 172, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Rygus, T.; Hillen, W. Inducible high-level expression of heterologous genes in Bacillus megaterium using the regulatory elements of the xylose-utilization operon. Appl. Microbiol. Biotechnol. 1991, 35, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Bjerga, G.E.K.; Uni Research, Bergen, Norway. RF-cloning using linear and circular plasmids as templates. 2016. [Google Scholar]
- Kim, L.; Mogk, A.; Schumann, W. A xylose-inducible Bacillus subtilis integration vector and its application. Gene 1996, 181, 71–76. [Google Scholar] [CrossRef]
- Ruiz, C.; Blanco, A.; Pastor, F.I.J.; Diaz, P. Analysis of Bacillus megaterium lipolytic system and cloning of LipA, a novel subfamily I.4 bacterial lipase. FEMS Microbiol. Lett. 2002, 217, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Feilmeier, B.J.; Iseminger, G.; Schroeder, D.; Webber, H.; Phillips, G.J. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J. Bacteriol. 2000, 182, 4068–4076. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Huang, Q.; Zhang, R.H.; Zhang, Y.Z. Purification and characterization of a fibrinolytic enzyme produced by Bacillus amyloliquefaciens DC-4 screened from douchi, a traditional Chinese soybean food. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2003, 134, 45–52. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, Ø.; Bjerga, G.E.K. Development of Versatile Vectors for Heterologous Expression in Bacillus. Microorganisms 2018, 6, 51. https://doi.org/10.3390/microorganisms6020051
Larsen Ø, Bjerga GEK. Development of Versatile Vectors for Heterologous Expression in Bacillus. Microorganisms. 2018; 6(2):51. https://doi.org/10.3390/microorganisms6020051
Chicago/Turabian StyleLarsen, Øivind, and Gro Elin Kjæreng Bjerga. 2018. "Development of Versatile Vectors for Heterologous Expression in Bacillus" Microorganisms 6, no. 2: 51. https://doi.org/10.3390/microorganisms6020051
APA StyleLarsen, Ø., & Bjerga, G. E. K. (2018). Development of Versatile Vectors for Heterologous Expression in Bacillus. Microorganisms, 6(2), 51. https://doi.org/10.3390/microorganisms6020051