Detection and Genetic Characterization of Puumala Orthohantavirus S-Segment in Areas of France Non-Endemic for Nephropathia Epidemica

 ,
,
Abstract
1. Introduction
2. Results
2.1. Seroprevalence
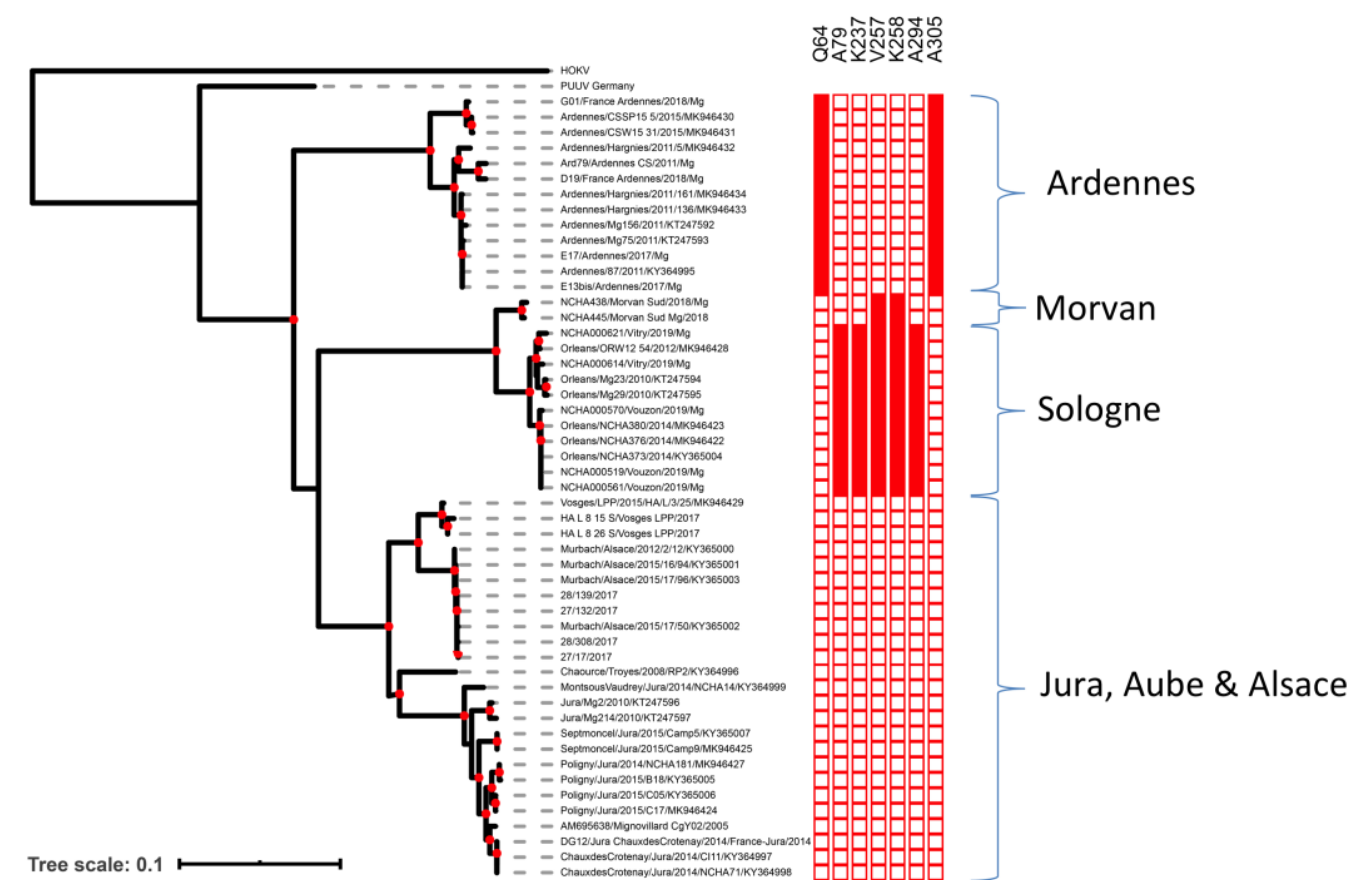
2.2. Phylogenetic Analyses and Identification of Clade-Specific Amino-Acid Signatures
2.3. Molecular Signatures of Selection
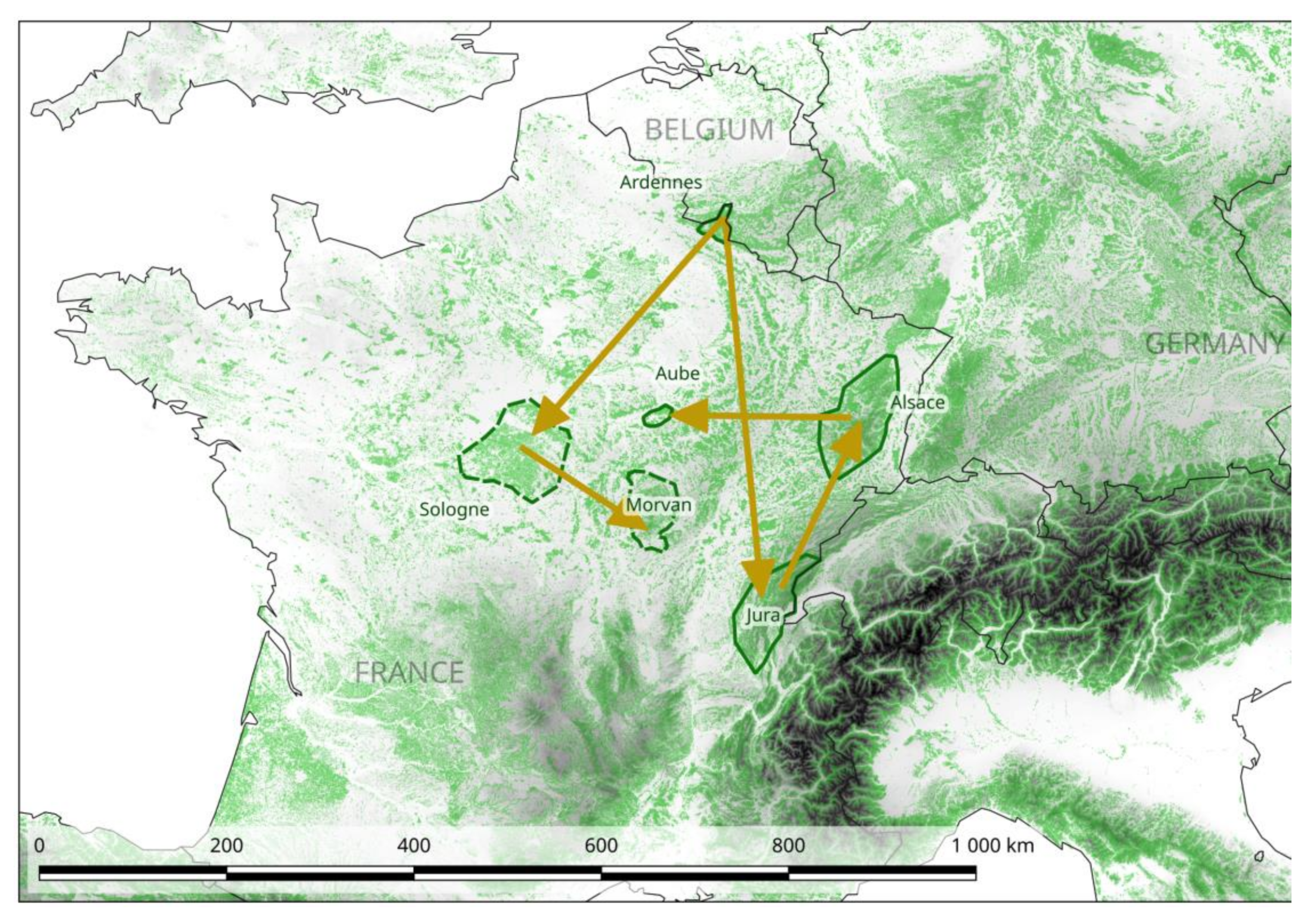
2.4. Phylogeography of PUUV at the National Scale in France
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Rodent Sampling
4.3. Serological and Virological Analyses
4.4. Phylogenetic Analyses and Detection of Clade-Specific Amino-Acid Signatures
4.5. Detection of Signatures of Selection
4.6. Estimation of Evolutionary Divergence
4.7. Phylogeographic Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- (ECDC) European Center for Disease Prevantion and Control. Facts about Hantavirus. Available online: https://www.ecdc.europa.eu/en/hantavirus-infection/facts (accessed on 1 June 2020).
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Jaaskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Lehvaslaiho, H.; Apekina, N.; Mikhailova, T.; Gavrilovskaya, I.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Brummer-Korvenkontio, M.; et al. Genetic variation of wild Puumala viruses within the serotype, local rodent populations and individual animal. Virus Res. 1995, 38, 25–41. [Google Scholar] [CrossRef]
- Brummer-Korvenkontio, M.; Henttonen, H.; Vaheri, A. Hemorrhagic fever with renal syndrome in Finland: Ecology and virology of nephropathia epidemica. Scand. J. Infect. Dis. Suppl. 1982, 36, 88–91. [Google Scholar] [PubMed]
- Bernshtein, A.D.; Apekina, N.S.; Mikhailova, T.V.; Myasnikov, Y.A.; Khlyap, L.A.; Korotkov, Y.S.; Gavrilovskaya, I.N. Dynamics of Puumala hantavirus infection in naturally infected bank voles (Clethrinomys glareolus). Arch. Virol. 1999, 144, 2415–2428. [Google Scholar] [CrossRef] [PubMed]
- Kallio, E.R.; Klingstrom, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, A. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, B.; Hornfeldt, B.; Lundkvist, A.; Bjorsten, S.; Leduc, J. Temporal dynamics of Puumala virus antibody prevalence in voles and of nephropathia epidemica incidence in humans. Am. J. Trop. Med. Hyg. 1995, 53, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Langlais, M.; Yoccoz, N.G.; Pontier, D. Modelling hantavirus in fluctuating populations of bank voles: The role of indirect transmission on virus persistence. J. Anim. Ecol. 2003, 72, 1–13. [Google Scholar] [CrossRef]
- Linard, C.; Tersago, K.; Leirs, H.; Lambin, E.F. Environmental conditions and Puumala virus transmission in Belgium. Int. J. Health Geogr. 2007, 6, 55. [Google Scholar] [CrossRef]
- Olsson, G.E.; Leirs, H.; Henttonen, H. Hantaviruses and their hosts in Europe: Reservoirs here and there, but not everywhere? Vector Borne Zoonotic Dis. 2010, 10, 549–561. [Google Scholar] [CrossRef]
- Quéré, J.P.; Le Louarn, H. Les Rongeurs de France: Faunistique et Biologie; Editions Quae: Paris, France, 2011; p. 311. [Google Scholar]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.F.; Plyusnin, A.; Marianneau, P.; et al. Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef] [PubMed]
- Monchatre-Leroy, E.; Crespin, L.; Boue, F.; Marianneau, P.; Calavas, D.; Henaux, V. Spatial and Temporal Epidemiology of Nephropathia Epidemica Incidence and Hantavirus Seroprevalence in Rodent Hosts: Identification of the Main Environmental Factors in Europe. Transbound. Emerg. Dis. 2017, 64, 1210–1228. [Google Scholar] [CrossRef] [PubMed]
- Reil, D.; Imholt, C.; Rosenfeld, U.M.; Drewes, S.; Fischer, S.; Heuser, E.; Petraityte-Burneikiene, R.; Ulrich, R.G.; Jacob, J. Validation of the Puumala virus rapid field test for bank voles in Germany. Epidemiol. Infect. 2017, 145, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Haredasht, S.A.; Taylor, C.J.; Maes, P.; Verstraeten, W.W.; Clement, J.; Barrios, M.; Lagrou, K.; Van Ranst, M.; Coppin, P.; Berckmans, D.; et al. Model-based prediction of nephropathia epidemica outbreaks based on climatological and vegetation data and bank vole population dynamics. Zoonoses Public Health 2013, 60, 461–477. [Google Scholar] [CrossRef]
- Tersago, K.; Verhagen, R.; Vapalahti, O.; Heyman, P.; Ducoffre, G.; Leirs, H. Hantavirus outbreak in Western Europe: Reservoir host infection dynamics related to human disease patterns. Epidemiol. Infect. 2011, 139, 381–390. [Google Scholar] [CrossRef]
- Reynes, J.M.; Dutrop, C.M.; Carli, D.; Levast, M.; Fontaine, N.; Denoyel, G.A.; Philit, J.B. Puumala hantavirus infection in Isere: Geographic extension of this zoonosis in France. Med. Mal. Infect. 2015, 45, 177–180. [Google Scholar] [CrossRef]
- National Reference Center for Hantaviruses. Surveillance Chez L’homme en France en 2019. Available online: https://www.pasteur.fr/fr/file/32257/download (accessed on 1 June 2020).
- Kruger, D.H.; Schonrich, G.; Klempa, B. Human pathogenic hantaviruses and prevention of infection. Hum. Vaccines 2011, 7, 685–693. [Google Scholar] [CrossRef]
- Johansson, P.; Olsson, G.E.; Low, H.T.; Bucht, G.; Ahlm, C.; Juto, P.; Elgh, F. Puumala hantavirus genetic variability in an endemic region (Northern Sweden). Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2008, 8, 286–296. [Google Scholar] [CrossRef]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kuhnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Vrancken, B.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef]
- Laramore, S.E.; Scarpa, J.; Laramore, C.R.; Lin, J. Virulence variation of white spot syndrome virus in Pacific white shrimp Litopenaeus vannamei. J. Aquat. Anim. Health 2009, 21, 82–90. [Google Scholar] [CrossRef]
- Feuer, R.; Boone, J.D.; Netski, D.; Morzunov, S.P.; St Jeor, S.C. Temporal and spatial analysis of Sin Nombre virus quasispecies in naturally infected rodents. J. Virol. 1999, 73, 9544–9554. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [PubMed]
- Weesendorp, E.; Stegeman, A.; Loeffen, W. Dynamics of virus excretion via different routes in pigs experimentally infected with classical swine fever virus strains of high, moderate or low virulence. Vet. Microbiol. 2009, 133, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Botha, E.M.; Markotter, W.; Wolfaardt, M.; Paweska, J.T.; Swanepoel, R.; Palacios, G.; Nel, L.H.; Venter, M. Genetic determinants of virulence in pathogenic lineage 2 West Nile virus strains. Emerg. Infect. Dis. 2008, 14, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, N.; Gonzalez-Scarano, F. Patterns of Infection-Unwanted Guests—Quick Visits and Extended Stays. In Viral Pathogenesis; Elsevier: Amsterdam, The Netherlands, 2016; pp. 81–94. [Google Scholar]
- Schrauwen, E.J.; de Graaf, M.; Herfst, S.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Determinants of virulence of influenza A virus. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2014, 33, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Hesse, R.R. Dengue Virus Evolution and Virulence Models. Clin. Infect. Dis. 2007, 44, 1462–1466. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Bernini, F.; Lo Presti, A.; Rezza, G.; Delogu, M.; Galli, M.; Ciccozzi, M. Phylogeography and epidemiological history of West Nile virus genotype 1a in Europe and the Mediterranean basin. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2011, 11, 646–653. [Google Scholar] [CrossRef]
- Pybus, O.G.; Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009, 10, 540–550. [Google Scholar] [CrossRef]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef]
- Reynes, J.M.; Carli, D.; Thomas, D.; Castel, G. Puumala Hantavirus Genotypes in Humans, France, 2012-2016. Emerg. Infect. Dis. 2019, 25, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Monchatre-Leroy, E.; Murri, S.; Castel, G.; Calavas, D.; Boue, F.; Henaux, V.; Marianneau, P. First insights into Puumala orthohantavirus circulation in a rodent population in Alsace, France. Zoonoses Public Health 2018, 65, 540–551. [Google Scholar] [CrossRef]
- Corine Land Cover (CLC). Available online: https://land.copernicus.eu/pan-european/corine-land-cover/clc-2012?tab=metadata. (accessed on 1 June 2020).
- Sadkowska-Todys, M.; Gut, W.; Baumann, A.; Siennicka, J.; Litwinska, B.; Zielinski, A. Occurrence of human hantavirus infections in Poland. Przegl Epidemiol. 2007, 61, 497–503. [Google Scholar]
- Krautkramer, E.; Grouls, S.; Urban, E.; Schnitzler, P.; Zeier, M. No gender-related differences in the severity of nephropathia epidemica, Germany. BMC Infect. Dis. 2013, 13, 457. [Google Scholar] [CrossRef] [PubMed]
- Wroblewska-Luczka, P.; Chmielewska-Badora, J.; Zwolinski, J.; Gaginskaya, A.R.; Adamczuk, P.; Zukiewicz-Sobczak, W.; Zagorski, J.; Wojtyla, A. Seroepidemiologic evaluation of exposure to infection with Hantavirus (serotype Puumala) among forestry workers in Poland. Balt. For. 2017, 23, 612–618. [Google Scholar]
- Engler, O.; Klingstrom, J.; Aliyev, E.; Niederhauser, C.; Fontana, S.; Strasser, M.; Portmann, J.; Signer, J.; Bankoul, S.; Frey, F.; et al. Seroprevalence of hantavirus infections in Switzerland in 2009: Difficulties in determining prevalence in a country with low endemicity. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. Eur. Commun. Dis. Bull. 2013, 18, 20660. [Google Scholar] [CrossRef] [PubMed]
- Ahlm, C.; Linderholm, M.; Juto, P.; Stegmayr, B.; Settergren, B. Prevalence of serum IgG antibodies to Puumala virus (haemorrhagic fever with renal syndrome) in northern Sweden. Epidemiol. Infect. 1994, 113, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Charbonnel, N.; Pages, M.; Sironen, T.; Henttonen, H.; Vapalahti, O.; Mustonen, J.; Vaheri, A. Immunogenetic factors affecting susceptibility of humans and rodents to hantaviruses and the clinical course of hantaviral disease in humans. Viruses 2014, 6, 2214–2241. [Google Scholar] [CrossRef]
- Dubois, A.; Castel, G.; Murri, S.; Pulido, C.; Pons, J.B.; Benoit, L.; Loiseau, A.; Lakhdar, L.; Galan, M.; Charbonnel, N.; et al. Experimental infections of wild bank voles (Myodes glareolus) from nephropatia epidemica endemic and non-endemic regions revealed slight differences in Puumala virological course and immunological responses. Virus Res. 2017, 235, 67–72. [Google Scholar] [CrossRef]
- Dubois, A.; Castel, G.; Murri, S.; Pulido, C.; Pons, J.B.; Benoit, L.; Loiseau, A.; Lakhdar, L.; Galan, M.; Marianneau, P.; et al. Bank vole immunoheterogeneity may limit Nephropatia Epidemica emergence in a French non-endemic region. Parasitology 2018, 145, 393–407. [Google Scholar] [CrossRef]
- Weesendorp, E.; Stegeman, A.; Loeffen, W.L. Quantification of classical swine fever virus in aerosols originating from pigs infected with strains of high, moderate or low virulence. Vet. Microbiol. 2009, 135, 222–230. [Google Scholar] [CrossRef]
- Horling, J.; Cheng, Y.; Plyusnin, A.; Persson, K.; Lehvaslaiho, H.; Vaheri, A.; Niklasson, B.; Lundkvist, A. Nucleotide and deduced amino acid sequences of the M and S genome segments of a Swedish Puumala virus isolate. Virus Res. 1995, 39, 321–330. [Google Scholar] [CrossRef]
- Klempa, B.; Tkachenko, E.A.; Dzagurova, T.K.; Yunicheva, Y.V.; Morozov, V.G.; Okulova, N.M.; Slyusareva, G.P.; Smirnov, A.; Kruger, D.H. Hemorrhagic fever with renal syndrome caused by 2 lineages of Dobrava hantavirus, Russia. Emerg. Infect. Dis. 2008, 14, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Garanina, E.; Martynova, E.; Davidyuk, Y.; Kabwe, E.; Ivanov, K.; Titova, A.; Markelova, M.; Zhuravleva, M.; Cherepnev, G.; Shakirova, V.G.; et al. Cytokine Storm Combined with Humoral Immune Response Defect in Fatal Hemorrhagic Fever with Renal Syndrome Case, Tatarstan, Russia. Viruses 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Klingstrom, J.; Smed-Sorensen, A.; Maleki, K.T.; Sola-Riera, C.; Ahlm, C.; Bjorkstrom, N.K.; Ljunggren, H.G. Innate and adaptive immune responses against human Puumala virus infection: immunopathogenesis and suggestions for novel treatment strategies for severe hantavirus-associated syndromes. J. Intern. Med. 2019, 285, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Rang, A.; Lutteke, N.; Raftery, M.J.; Charbonnel, N.; Ulrich, R.G. Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 2008, 225, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Kallio-Kokko, H.; Narvanen, A.; Julkunen, I.; Lundkvist, A.; Plyusnin, A.; Lehvaslaiho, H.; Brummer-Korvenkontio, M.; Vaheri, A.; Lankinen, H. Human B-cell epitopes of Puumala virus nucleocapsid protein, the major antigen in early serological response. J. Med. Virol. 1995, 46, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Elgh, F.; Lundkvist, A.; Alexeyev, O.A.; Wadell, G.; Juto, P. A major antigenic domain for the human humoral response to Puumala virus nucleocapsid protein is located at the amino-terminus. J. Virol. Methods 1996, 59, 161–172. [Google Scholar] [CrossRef]
- Lundkvist, A.; Kallio-Kokko, H.; Sjolander, K.B.; Lankinen, H.; Niklasson, B.; Vaheri, A.; Vapalahti, O. Characterization of Puumala virus nucleocapsid protein: Identification of B-cell epitopes and domains involved in protective immunity. Virology 1996, 216, 397–406. [Google Scholar] [CrossRef]
- Gott, P.; Zoller, L.; Darai, G.; Bautz, E.K. A major antigenic domain of hantaviruses is located on the aminoproximal site of the viral nucleocapsid protein. Virus Genes 1997, 14, 31–40. [Google Scholar] [CrossRef]
- De Carvalho Nicacio, C.; Gonzalez Della Valle, M.; Padula, P.; Bjorling, E.; Plyusnin, A.; Lundkvist, A. Cross-protection against challenge with Puumala virus after immunization with nucleocapsid proteins from different hantaviruses. J. Virol. 2002, 76, 6669–6677. [Google Scholar] [CrossRef]
- De Carvalho Nicacio, C.; Sallberg, M.; Hultgren, C.; Lundkvist, A. T-helper and humoral responses to Puumala hantavirus nucleocapsid protein: Identification of T-helper epitopes in a mouse model. J. Gen. Virol. 2001, 82, 129–138. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Holmes, E.C. The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 2018, 19, 756–769. [Google Scholar] [CrossRef]
- Pilaski, J.; Feldmann, H.; Morzunov, S.; Rollin, P.E.; Ruo, S.L.; Lauer, B.; Peters, C.J.; Nichol, S.T. Genetic identification of a new Puumala virus strain causing severe hemorrhagic fever with renal syndrome in Germany. J. Infect. Dis. 1994, 170, 1456–1462. [Google Scholar] [CrossRef]
- Sironen, T.; Kallio, E.R.; Vaheri, A.; Lundkvist, A.; Plyusnin, A. Quasispecies dynamics and fixation of a synonymous mutation in hantavirus transmission. J. Gen. Virol. 2008, 89, 1309–1313. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Gunthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef]
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Cyclic hantavirus epidemics in humans--predicted by rodent host dynamics. Epidemics 2009, 1, 101–107. [Google Scholar] [CrossRef]
- Tagliapietra, V.; Rosa, R.; Rossi, C.; Rosso, F.; Hauffe, H.C.; Tommasini, M.; Versini, W.; Cristallo, A.F.; Rizzoli, A. Emerging Rodent-Borne Viral Zoonoses in Trento, Italy. EcoHealth 2018, 15, 695–704. [Google Scholar] [CrossRef]
- Clement, J.; Maes, P.; van Ypersele de Strihou, C.; van der Groen, G.; Barrios, J.M.; Verstraeten, W.W.; van Ranst, M. Beechnuts and outbreaks of nephropathia epidemica (NE): Of mast, mice and men. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2010, 25, 1740–1746. [Google Scholar] [CrossRef]
- Zeimes, C.B.; Quoilin, S.; Henttonen, H.; Lyytikainen, O.; Vapalahti, O.; Reynes, J.M.; Reusken, C.; Swart, A.N.; Vainio, K.; Hjertqvist, M.; et al. Landscape and regional environmental analysis of the spatial distribution of hantavirus human cases in europe. Front. Public Health 2015, 3, 54. [Google Scholar] [CrossRef]
- Zeimes, C.B.; Olsson, G.E.; Ahlm, C.; Vanwambeke, S.O. Modelling zoonotic diseases in humans: Comparison of methods for hantavirus in Sweden. Int. J. Health Geogr. 2012, 11, 39. [Google Scholar] [CrossRef]
- Godefroid, M.; Rasplus, J.-Y.; Rossi, J.-P. Is phylogeography helpful for invasive species risk assessment? The case study of the bark beetle genus Dendroctonus. Ecography 2016, 39, 1197–1209. [Google Scholar] [CrossRef]
- Dellicour, S.; Rose, R.; Pybus, O.G. Explaining the geographic spread of emerging epidemics: A framework for comparing viral phylogenies and environmental landscape data. BMC Bioinform. 2016, 17, 82. [Google Scholar] [CrossRef]
- Duchatel, F.; Bronsvoort, B.M.d.C.; Lycett, S. Phylogeographic Analysis and Identification of Factors Impacting the Diffusion of Foot-and-Mouth Disease Virus in Africa. Front. Ecol. Evol. 2019, 7. [Google Scholar] [CrossRef]
- Tabachnick, W.J. Climate Change and the Arboviruses: Lessons from the Evolution of the Dengue and Yellow Fever Viruses. Annu. Rev. Virol. 2016, 3, 125–145. [Google Scholar] [CrossRef]
- Lu, L.; Leigh Brown, A.J.; Lycett, S.J. Quantifying predictors for the spatial diffusion of avian influenza virus in China. BMC Evol. Biol. 2017, 17, 16. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef]
- Faber, M.; Kruger, D.H.; Auste, B.; Stark, K.; Hofmann, J.; Weiss, S. Molecular and epidemiological characteristics of human Puumala and Dobrava-Belgrade hantavirus infections, Germany, 2001 to 2017. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. Eur. Commun. Dis. Bull. 2019, 24. [Google Scholar] [CrossRef]
- Duchene, S.; Lanfear, R. Phylogenetic uncertainty can bias the number of evolutionary transitions estimated from ancestral state reconstruction methods. J. Exp. Zool. Part B Mol. Dev. Evol. 2015, 324, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Laenen, L.; Dellicour, S.; Vergote, V.; Nauwelaers, I.; De Coster, S.; Verbeeck, I.; Vanmechelen, B.; Lemey, P.; Maes, P. Spatio-temporal analysis of Nova virus, a divergent hantavirus circulating in the European mole in Belgium. Mol. Ecol. 2016, 25, 5994–6008. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Kosakovsky Pond, S.L. Purifying selection can obscure the ancient age of viral lineages. Mol. Biol. Evol. 2011, 28, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef]
- Dambrine, E.; Dupouey, J.L.; Laut, L.; Humbert, L.; Thinon, M.; Beaufils, T.; Richard, H. Present forest biodiversity patterns in france related to former Roman agriculture. Ecology 2007, 88, 1430–1439. [Google Scholar] [CrossRef]
- Vallauri, D.; Grel, A.; Granier, E.; Dupouey, J.L. Les Forets de Cassini. Analyse Quantitative et Comparaison Avec Les Forêts Actuelles; WWF/INRA: Marseilles, France, 2012; p. 64. [Google Scholar]
- Dupouey, J.L.; Dambrine, E.; Laffite, J.D.; Moares, C. Irreversible impact of past land use on forest soils and biodiversity. Ecology 2002, 83, 2978–2984. [Google Scholar] [CrossRef]
- Klingstrom, J.; Heyman, P.; Escutenaire, S.; Sjolander, K.B.; De Jaegere, F.; Henttonen, H.; Lundkvist, A. Rodent host specificity of European hantaviruses: Evidence of Puumala virus interspecific spillover. J. Med. Virol. 2002, 68, 581–588. [Google Scholar] [CrossRef]
- Hardestam, J.; Karlsson, M.; Falk, K.I.; Olsson, G.; Klingstrom, J.; Lundkvist, A. Puumala hantavirus excretion kinetics in bank voles (Myodes glareolus). Emerg. Infect. Dis. 2008, 14, 1209–1215. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree, 1.4; 2009. Institute of Evolutionary Biology, University of Edinburgh. Available online: http://tree.bio.ed.ac.uk/ (accessed on 1 June 2020).
- Korber, B.; Myers, G. Signature pattern analysis: A method for assessing viral sequence relatedness. AIDS Res. Hum. Retrovir. 1992, 8, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef]
- Spielman, S.J.; Weaver, S.; Shank, S.D.; Magalis, B.R.; Li, M.; Kosakovsky Pond, S.L. Evolution of Viral Genomes: Interplay Between Selection, Recombination, and Other Forces. Methods Mol. Biol. 2019, 1910, 427–468. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P. Bayesian evolutionary model testing in the phylogenomics era: Matching model complexity with computational efficiency. Bioinformatics 2013, 29, 1970–1979. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Chevenet, F.; Castel, G.; Jousselin, E.; Gascuel, O. PastView: A user-friendly interface to explore evolutionary scenarios. bioRxiv 2019. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Geographic Coordinates | Forest | Sampling Period | Number Seropositive for PUUV/Number of Bank Voles Trapped | Number of Seropositive Bank Voles That Are Positive in qRT-PCR |
|---|---|---|---|---|---|
| Roussillon-en-Morvan | 47.01° N, 4.07° E | Morvan (South) | September 2018 | 3/10 | 3/3 |
| Avallon/Vézelay | 47.47° N, 3.86° E | Morvan (North) | September 2018 | 0/2 | - |
| Tronçais | 46.66° N, 2.74° E | Tronçais | September 2019 | 0/62 | - |
| Vouzon | 47.66° N, 2.10° E | Sologne (South) | October 2019 | 17/65 | 17/17 |
| Vitry aux Loges | 47.96° N, 2.26° E | Sologne (North-East) | October 2019 | 10/70 | 10/10 |
| Ingrannes | 47.94° N, 2.21° E | Sologne (North-West) | October 2018 | 0/45 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murri, S.; Madrières, S.; Tatard, C.; Piry, S.; Benoit, L.; Loiseau, A.; Pradel, J.; Artige, E.; Audiot, P.; Leménager, N.; et al. Detection and Genetic Characterization of Puumala Orthohantavirus S-Segment in Areas of France Non-Endemic for Nephropathia Epidemica. Pathogens 2020, 9, 721. https://doi.org/10.3390/pathogens9090721
Murri S, Madrières S, Tatard C, Piry S, Benoit L, Loiseau A, Pradel J, Artige E, Audiot P, Leménager N, et al. Detection and Genetic Characterization of Puumala Orthohantavirus S-Segment in Areas of France Non-Endemic for Nephropathia Epidemica. Pathogens. 2020; 9(9):721. https://doi.org/10.3390/pathogens9090721
Chicago/Turabian StyleMurri, Séverine, Sarah Madrières, Caroline Tatard, Sylvain Piry, Laure Benoit, Anne Loiseau, Julien Pradel, Emmanuelle Artige, Philippe Audiot, Nicolas Leménager, and et al. 2020. "Detection and Genetic Characterization of Puumala Orthohantavirus S-Segment in Areas of France Non-Endemic for Nephropathia Epidemica" Pathogens 9, no. 9: 721. https://doi.org/10.3390/pathogens9090721
APA StyleMurri, S., Madrières, S., Tatard, C., Piry, S., Benoit, L., Loiseau, A., Pradel, J., Artige, E., Audiot, P., Leménager, N., Lacôte, S., Vulin, J., Charbonnel, N., Marianneau, P., & Castel, G. (2020). Detection and Genetic Characterization of Puumala Orthohantavirus S-Segment in Areas of France Non-Endemic for Nephropathia Epidemica. Pathogens, 9(9), 721. https://doi.org/10.3390/pathogens9090721