The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD)

Abstract
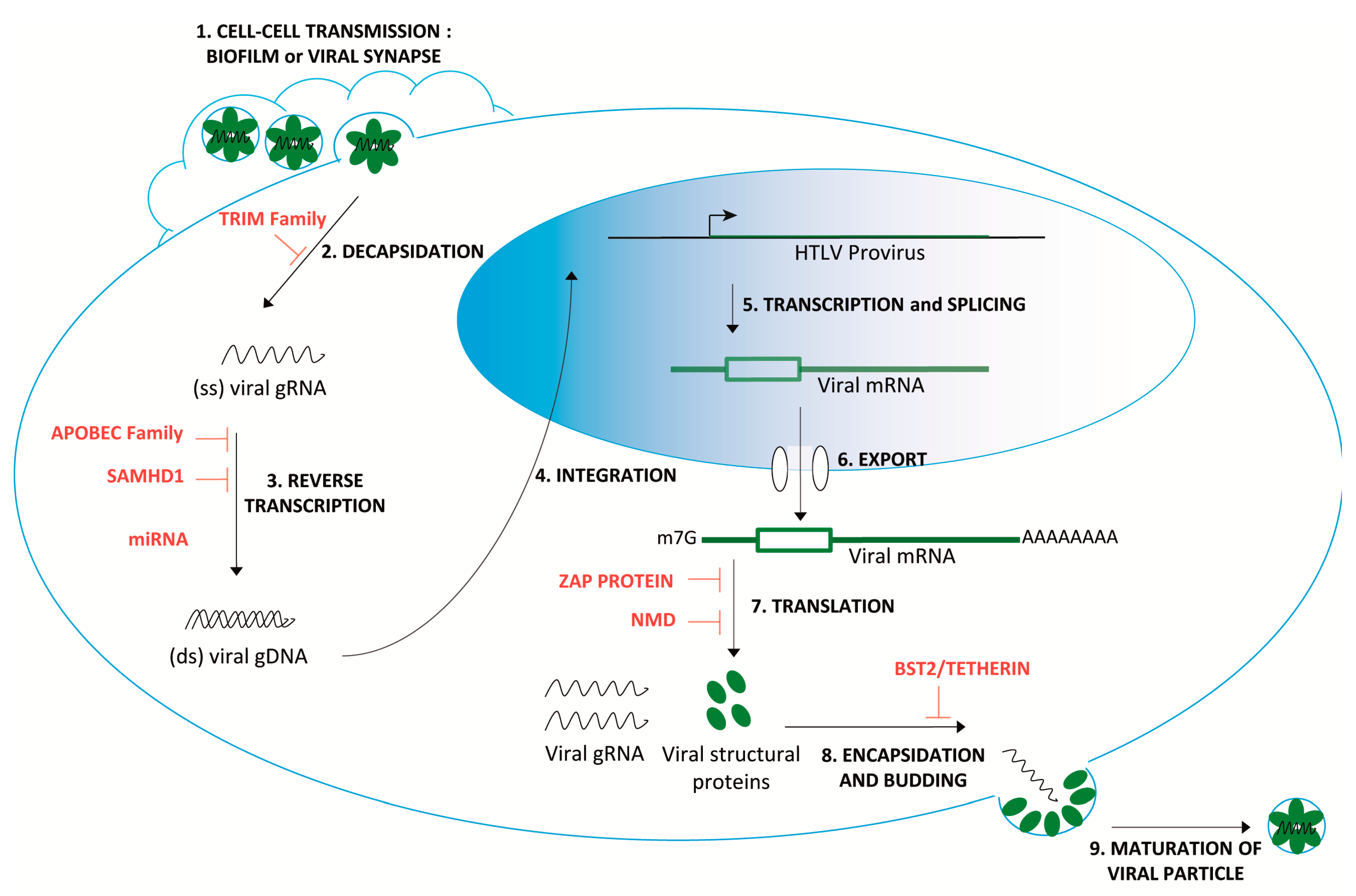
1. Introduction
2. NMD in the Cell
2.1. Mechanism and Regulation
2.2. NMD Targets and Functions
3. HTLV-1 Confronts NMD
3.1. HTLV-1 Viral mRNA Exhibits NMD-Initiating Features
3.2. HTLV-1 Protection against NMD
3.3. When Does NMD Inhibition Occur during HTLV-1 Infection?
3.4. HTLV-1-Associated Pathologies. How Can NMD Inhibition Impact the Host in the Long Term?
4. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviation
| HTLV-1 | Human T cell leukaemia/lymphoma virus type 1. |
| ATL | Adult T cell leukemia/lymphoma. |
| HAM/TSP | HTLV-1 associated myelopathy/ tropical spastic paraparesis. |
| TRIM | Tripartite motif-containing protein. |
| APOBEC | Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like. |
| SAMHD1 | SAM domain and HD domain-containing protein 1. |
| BTS2 | Bone marrow stromal cell antigen 2. |
| ZAP | Zinc finger Antiviral Protein. |
| NMD | nonsense-mediated mRNA decay. |
| mRNP | Ribonucleoprotein associated with mRNA. |
| UTR | untranslated region. |
| PTC | Premature termination codon. |
| EJC | Exon-junction complex. |
| SMG1 PI3K | SMG1 nonsense mediated mRNA decay associated PI3K related kinase. |
| CCR4-NOT | The multi-subunit CCR4 (carbon catabolite repressor 4)-NOT (Negative on TATA) complex. |
| DCP1a/2 | mRNA-decapping enzyme subunit 1a/2. |
| CREB-2/ATF4 | C-AMP Response Element-binding protein 2/Activating transcription factor 4. |
References
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Enose-Akahata, Y.; Vellucci, A.; Jacobson, S. Role of HTLV-1 Tax and HBZ in the Pathogenesis of HAM/TSP. Front. Microbiol. 2017, 8, 2563. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.M. Human T Cell Leukemia Virus Type 1: Persistence and Pathogenesis. Annu. Rev. Immunol. 2018, 36, 43–71. [Google Scholar] [CrossRef]
- Alais, S.; Mahieux, R.; Dutartre, H. Viral Source-Independent High Susceptibility of Dendritic Cells to Human T-Cell Leukemia Virus Type 1 Infection Compared to That of T Lymphocytes. J. Virol. 2015, 89, 10580–10590. [Google Scholar] [CrossRef]
- Furuta, R.; Yasunaga, J.; Miura, M.; Sugata, K.; Saito, A.; Akari, H.; Ueno, T.; Takenouchi, N.; Fujisawa, J.; Koh, K.-R.; et al. Human T-cell leukemia virus type 1 infects multiple lineage hematopoietic cells in vivo. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Itoyama, Y.; Nakamura, N.; Takamatsu, K.; Kira, J.; Iwamasa, T.; Goto, I.; Yamamoto, N. In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology 1993, 196, 25–33. [Google Scholar] [CrossRef]
- Bangham, C.R.M. CTL quality and the control of human retroviral infections. Eur. J. Immunol. 2009, 39, 1700–1712. [Google Scholar] [CrossRef]
- Gazon, H.; Chauhan, P.; Hamaidia, M.; Hoyos, C.; Li, L.; Safari, R.; Willems, L. How Does HTLV-1 Undergo Oncogene-Dependent Replication Despite a Strong Immune Response? Front. Microbiol. 2017, 8, 2684. [Google Scholar] [CrossRef]
- Rocamonde, B.; Carcone, A.; Mahieux, R.; Dutartre, H. HTLV-1 infection of myeloid cells: From transmission to immune alterations. Retrovirology 2019, 16, 45. [Google Scholar] [CrossRef]
- Forlani, G.; Shallak, M.; Ramia, E.; Tedeschi, A.; Accolla, R.S. Restriction factors in human retrovirus infections and the unprecedented case of CIITA as link of intrinsic and adaptive immunity against HTLV-1. Retrovirology 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Maldonado, J.O.; Mueller, J.D.; Zhang, W.; Mansky, L.M. Molecular Studies of HTLV-1 Replication: An Update. Viruses 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Nozuma, S.; Matsuura, E.; Kodama, D.; Tashiro, Y.; Matsuzaki, T.; Kubota, R.; Izumo, S.; Takashima, H. Effects of host restriction factors and the HTLV-1 subtype on susceptibility to HTLV-1-associated myelopathy/tropical spastic paraparesis. Retrovirology 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Leal, F.E.; Menezes, S.M.; Costa, E.A.S.; Brailey, P.M.; Gama, L.; Segurado, A.C.; Kallas, E.G.; Nixon, D.F.; Dierckx, T.; Khouri, R.; et al. Comprehensive Antiretroviral Restriction Factor Profiling Reveals the Evolutionary Imprint of the ex Vivo and in Vivo IFN-β Response in HTLV-1-Associated Neuroinflammation. Front. Microbiol. 2018, 9, 985. [Google Scholar] [CrossRef]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Müllers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of diverse retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef]
- Chiang, K.; Rice, A.P. MicroRNA-mediated restriction of HIV-1 in resting CD4+ T cells and monocytes. Viruses 2012, 4, 1390–1409. [Google Scholar] [CrossRef]
- Sampey, G.C.; Van Duyne, R.; Currer, R.; Das, R.; Narayanan, A.; Kashanchi, F. Complex role of microRNAs in HTLV-1 infections. Front. Genet. 2012, 3, 295. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, X.; Goff, S.P.; Gao, G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012, 31, 4236–4246. [Google Scholar] [CrossRef]
- Soliman, M.; Srikrishna, G.; Balagopal, A. Mechanisms of HIV Control. Curr. HIV/AIDS Rep. 2017, 14, 101–109. [Google Scholar] [CrossRef]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; de Thé, H.; Bazarbachi, A. ATL response to arsenic/interferon therapy is triggered by SUMO/PML/RNF4-dependent Tax degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef]
- Mahieux, R.; Suspène, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.-P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Sasada, A.; Takaori-Kondo, A.; Shirakawa, K.; Kobayashi, M.; Abudu, A.; Hishizawa, M.; Imada, K.; Tanaka, Y.; Uchiyama, T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2005, 2, 32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ooms, M.; Krikoni, A.; Kress, A.K.; Simon, V.; Münk, C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012, 86, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.P.; Matsuoka, M. APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.-I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Yao, J.; Tanaka, M.; Takenouchi, N.; Ren, Y.; Lee, S.-I.; Fujisawa, J.-I. Induction of APOBEC3B cytidine deaminase in HTLV-1-infected humanized mice. Exp. Ther. Med. 2019, 17, 3701–3708. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef]
- Bai, X.T.; Nicot, C. miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 2015, 290, 5381–5390. [Google Scholar] [CrossRef]
- Ilinskaya, A.; Derse, D.; Hill, S.; Princler, G.; Heidecker, G. Cell-cell transmission allows human T-lymphotropic virus 1 to circumvent tetherin restriction. Virology 2013, 436, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Miyazato, P.; Matsuo, M.; Tan, B.J.Y.; Tokunaga, M.; Katsuya, H.; Islam, S.; Ito, J.; Murakawa, Y.; Satou, Y. HTLV-1 contains a high CG dinucleotide content and is susceptible to the host antiviral protein ZAP. Retrovirology 2019, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.J.; Beemon, K.L. Unspliced Rous sarcoma virus genomic RNAs are translated and subjected to nonsense-mediated mRNA decay before packaging. J. Virol. 2004, 78, 5139–5146. [Google Scholar] [CrossRef]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular NMD Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. mBio 2018, 9. [Google Scholar] [CrossRef]
- Li, M.; Johnson, J.R.; Truong, B.; Kim, G.; Weinbren, N.; Dittmar, M.; Shah, P.S.; Von Dollen, J.; Newton, B.W.; Jang, G.M.; et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol. 2019, 4, 985–995. [Google Scholar] [CrossRef]
- Garcia, D.; Garcia, S.; Voinnet, O. Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 2014, 16, 391–402. [Google Scholar] [CrossRef]
- Rao, S.; Amorim, R.; Niu, M.; Breton, Y.; Tremblay, M.J.; Mouland, A.J. Host mRNA decay proteins influence HIV-1 replication and viral gene expression in primary monocyte-derived macrophages. Retrovirology 2019, 16, 3. [Google Scholar] [CrossRef]
- Wada, M.; Lokugamage, K.G.; Nakagawa, K.; Narayanan, K.; Makino, S. Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10157–E10166. [Google Scholar] [CrossRef]
- May, J.P.; Yuan, X.; Sawicki, E.; Simon, A.E. RNA virus evasion of nonsense-mediated decay. PLoS Pathog. 2018, 14, e1007459. [Google Scholar] [CrossRef]
- Lukhovitskaya, N.; Ryabova, L.A. Cauliflower mosaic virus transactivator protein (TAV) can suppress nonsense-mediated decay by targeting VARICOSE, a scaffold protein of the decapping complex. Sci. Rep. 2019, 9, 7042. [Google Scholar] [CrossRef]
- Mocquet, V.; Neusiedler, J.; Rende, F.; Cluet, D.; Robin, J.P.; Terme, J.M.; Duc Dodon, M.; Wittmann, J.; Morris, C.; Le Hir, H.; et al. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J. Virol. 2012, 86, 7530–7543. [Google Scholar] [CrossRef]
- Nakano, K.; Ando, T.; Yamagishi, M.; Yokoyama, K.; Ishida, T.; Ohsugi, T.; Tanaka, Y.; Brighty, D.W.; Watanabe, T. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: Implications for retroviral replication. Microbes Infect. 2013, 15, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Zund, D.; Gruber, A.R.; Zavolan, M.; Muhlemann, O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3’ UTRs. Nat. Struct. Mol. Biol. 2013, 20, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Hurt, J.A.; Robertson, A.D.; Burge, C.B. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013, 23, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C.; et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef] [PubMed]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef]
- Yamashita, A.; Izumi, N.; Kashima, I.; Ohnishi, T.; Saari, B.; Katsuhata, Y.; Muramatsu, R.; Morita, T.; Iwamatsu, A.; Hachiya, T.; et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 2009, 23, 1091–1105. [Google Scholar] [CrossRef]
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001, 15, 2215–2228. [Google Scholar] [CrossRef]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef]
- Hug, N.; Cáceres, J.F. The RNA helicase DHX34 activates NMD by promoting a transition from the surveillance to the decay-inducing complex. Cell Rep. 2014, 8, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Hug, N.; López-Perrote, A.; Yamashita, A.; Cáceres, J.F.; Llorca, O. The RNA helicase DHX34 functions as a scaffold for SMG1-mediated UPF1 phosphorylation. Nat. Commun. 2016, 7, 10585. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Uchiyama, A.; Castaño, R.; Kataoka, N.; Kurosawa, H.; Ohno, S.; Yamashita, A.; Llorca, O. Structures of SMG1-UPFs Complexes: SMG1 Contributes to Regulate UPF2-Dependent Activation of UPF1 in NMD. Structure 2014, 22, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Buchwald, G.; Castano, R.; Raabe, M.; Gil, D.; Lazaro, M.; Urlaub, H.; Conti, E.; Llorca, O. The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3’ end. Nat. Struct. Mol. Biol. 2012, 19, 498–505. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 2011, 41, 693–703. [Google Scholar] [CrossRef]
- Durand, S.; Franks, T.M.; Lykke-Andersen, J. Hyperphosphorylation amplifies UPF1 activity to resolve stalls in nonsense-mediated mRNA decay. Nat. Commun. 2016, 7, 12434. [Google Scholar] [CrossRef]
- Franks, T.M.; Singh, G.; Lykke-Andersen, J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 2010, 143, 938–950. [Google Scholar] [CrossRef]
- Cho, H.; Han, S.; Choe, J.; Park, S.G.; Choi, S.S.; Kim, Y.K. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res. 2013, 41, 1319–1328. [Google Scholar] [CrossRef]
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138. [Google Scholar] [CrossRef]
- Unterholzner, L.; Izaurralde, E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 2004, 16, 587–596. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.R.; Grimson, A.; Hachiya, T.; Hentze, M.W.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef]
- Dyle, M.C.; Kolakada, D.; Cortazar, M.A.; Jagannathan, S. How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip Rev. RNA 2020, 11, e1560. [Google Scholar] [CrossRef] [PubMed]
- Kishor, A.; Fritz, S.E.; Hogg, J.R. Nonsense-mediated mRNA decay: The challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip Rev. RNA 2019, 10, e1548. [Google Scholar] [CrossRef] [PubMed]
- Karousis, E.D.; Mühlemann, O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Kashima, I.; Jonas, S.; Jayachandran, U.; Buchwald, G.; Conti, E.; Lupas, A.N.; Izaurralde, E. SMG6 interacts with the exon junction complex via two conserved EJC-binding motifs (EBMs) required for nonsense-mediated mRNA decay. Genes Dev. 2010, 24, 2440–2450. [Google Scholar] [CrossRef]
- Mabin, J.W.; Woodward, L.A.; Patton, R.D.; Yi, Z.; Jia, M.; Wysocki, V.H.; Bundschuh, R.; Singh, G. The Exon Junction Complex Undergoes a Compositional Switch that Alters mRNP Structure and Nonsense-Mediated mRNA Decay Activity. Cell Rep. 2018, 25, 2431–2446.e7. [Google Scholar] [CrossRef]
- Ge, Z.; Quek, B.L.; Beemon, K.L.; Hogg, J.R. Polypyrimidine tract binding protein 1 protects mRNAs from recognition by the nonsense-mediated mRNA decay pathway. Elife 2016, 5. [Google Scholar] [CrossRef]
- Kishor, A.; Ge, Z.; Hogg, J.R. hnRNP L-dependent protection of normal mRNAs from NMD subverts quality control in B cell lymphoma. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Gardner, L.B. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell. Biol. 2008, 28, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T.; Sharifi, N.A.; Meyers, J.L.; Martinez-Murillo, F.; Dietz, H.C. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 2004, 36, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Porse, B.T. The functional consequences of intron retention: Alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 2014, 36, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef]
- Saltzman, A.L.; Kim, Y.K.; Pan, Q.; Fagnani, M.M.; Maquat, L.E.; Blencowe, B.J. Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol. Cell. Biol. 2008, 28, 4320–4330. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Waage, J.; Tian, G.; Zhao, J.; Damgaard, I.; Jakobsen, J.S.; Kristiansen, K.; Krogh, A.; Wang, J.; Porse, B.T. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012, 13, R35. [Google Scholar] [CrossRef]
- Belew, A.T.; Meskauskas, A.; Musalgaonkar, S.; Advani, V.M.; Sulima, S.O.; Kasprzak, W.K.; Shapiro, B.A.; Dinman, J.D. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 2014, 512, 265–269. [Google Scholar] [CrossRef]
- Mocquet, V.; Durand, S.; Jalinot, P. How retroviruses escape the Nonsense Mediated mRNA Decay (NMD)? AIDS Res. Hum. Retroviruses 2015. [Google Scholar] [CrossRef]
- Mazumder, B.; Seshadri, V.; Fox, P.L. Translational control by the 3’-UTR: The ends specify the means. Trends Biochem. Sci. 2003, 28, 91–98. [Google Scholar] [CrossRef]
- Weil, J.E.; Beemon, K.L. A 3’ UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA 2006, 12, 102–110. [Google Scholar] [CrossRef]
- Hogg, J.R.; Goff, S.P. Upf1 senses 3’UTR length to potentiate mRNA decay. Cell 2010, 143, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Giam, C.Z. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc. Natl. Acad. Sci. USA 1992, 89, 7070–7074. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Shao, S.; Murray, J.; Hegde, R.S.; Ramakrishnan, V. Structural basis for stop codon recognition in eukaryotes. Nature 2015, 524, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-L.; Wei, J.-Y.; Wang, L.; Huang, S.; Chen, J.-L. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharmacol. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Desbois, C.; Rousset, R.; Bantignies, F.; Jalinot, P. Exclusion of Int-6 from PML nuclear bodies by binding to the HTLV-I Tax oncoprotein. Science 1996, 273, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Wittmann, J.; Jack, H.M.; Jalinot, P. Human INT6/eIF3e is required for nonsense-mediated mRNA decay. EMBO Rep 2007, 8, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, F.; Robin, J.-P.; Kanaan, J.; Borowiak, M.; Croquette, V.; Le Hir, H.; Jalinot, P.; Mocquet, V. HTLV-1 Tax plugs and freezes UPF1 helicase leading to nonsense-mediated mRNA decay inhibition. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hidaka, M.; Inoue, J.; Yoshida, M.; Seiki, M. Post-transcriptional regulator (rex) of HTLV-1 initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO J. 1988, 7, 519–523. [Google Scholar] [CrossRef]
- Hakata, Y.; Umemoto, T.; Matsushita, S.; Shida, H. Involvement of human CRM1 (exportin 1) in the export and multimerization of the Rex protein of human T-cell leukemia virus type 1. J. Virol. 1998, 72, 6602–6607. [Google Scholar] [CrossRef]
- Nakano, K.; Watanabe, T. HTLV-1 Rex Tunes the Cellular Environment Favorable for Viral Replication. Viruses 2016, 8, 58. [Google Scholar] [CrossRef]
- Rao, S.; Amorim, R.; Niu, M.; Temzi, A.; Mouland, A.J. The RNA surveillance proteins UPF1, UPF2 and SMG6 affect HIV-1 reactivation at a post-transcriptional level. Retrovirology 2018, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Rende, F.; Cavallari, I.; Corradin, A.; Silic-Benussi, M.; Toulza, F.; Toffolo, G.M.; Tanaka, Y.; Jacobson, S.; Taylor, G.P.; D’Agostino, D.M.; et al. Kinetics and intracellular compartmentalization of HTLV-1 gene expression: Nuclear retention of HBZ mRNAs. Blood 2011, 117, 4855–4859. [Google Scholar] [CrossRef] [PubMed]
- Billman, M.R.; Rueda, D.; Bangham, C.R.M. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res. 2017, 2, 87. [Google Scholar] [CrossRef] [PubMed]
- Rowan, A.G.; Suemori, K.; Fujiwara, H.; Yasukawa, M.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R.M. Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of Tax expression. Retrovirology 2014, 11, 116. [Google Scholar] [CrossRef]
- Matsuoka, M.; Mesnard, J.-M. HTLV-1 bZIP factor: The key viral gene for pathogenesis. Retrovirology 2020, 17, 2. [Google Scholar] [CrossRef]
- Ho, Y.-K.; Zhi, H.; DeBiaso, D.; Philip, S.; Shih, H.-M.; Giam, C.-Z. HTLV-1 tax-induced rapid senescence is driven by the transcriptional activity of NF-κB and depends on chronically activated IKKα and p65/RelA. J. Virol. 2012, 86, 9474–9483. [Google Scholar] [CrossRef]
- Philip, S.; Zahoor, M.A.; Zhi, H.; Ho, Y.-K.; Giam, C.-Z. Regulation of human T-lymphotropic virus type I latency and reactivation by HBZ and Rex. PLoS Pathog. 2014, 10, e1004040. [Google Scholar] [CrossRef]
- Zhi, H.; Yang, L.; Kuo, Y.-L.; Ho, Y.-K.; Shih, H.-M.; Giam, C.-Z. NF-κB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011, 7, e1002025. [Google Scholar] [CrossRef]
- Giam, C.-Z.; Semmes, O.J. HTLV-1 Infection and Adult T-Cell Leukemia/Lymphoma-A Tale of Two Proteins: Tax and HBZ. Viruses 2016, 8, 161. [Google Scholar] [CrossRef]
- Yamagishi, M.; Watanabe, T. Molecular hallmarks of adult T cell leukemia. Front. Microbiol. 2012, 3, 334. [Google Scholar] [CrossRef]
- Wittmann, J.; Hol, E.M.; Jack, H.M. hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol. Cell. Biol. 2006, 26, 1272–1287. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.H.; Shao, A.; Shum, E.Y.; Espinoza, J.L.; Huang, L.; Karam, R.; Wilkinson, M.F. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014, 6, 748–764. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Furlan, A.; Gonzalez-Hilarion, S.; Leroy, C.; Gruenert, D.C.; Tulasne, D.; Lejeune, F. Caspases shutdown nonsense-mediated mRNA decay during apoptosis. Cell Death Differ. 2015, 22, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Maquat, L.E. Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat. Commun. 2015, 6, 6632. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.A.; Durand, S.; Dasgupta, A.; Radonovich, M.; Cimarelli, A.; Brady, J.N.; Mahieux, R.; Pise-Masison, C.A. The transcription profile of Tax-3 is more similar to Tax-1 than Tax-2: Insights into HTLV-3 potential leukemogenic properties. PLoS ONE 2012, 7, e41003. [Google Scholar] [CrossRef]
- Nelson, J.O.; Moore, K.A.; Chapin, A.; Hollien, J.; Metzstein, M.M. Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. Elife 2016, 5. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Damgaard, I.; Bryder, D.; Theilgaard-Monch, K.; Thoren, L.A.; Nielsen, F.C.; Jacobsen, S.E.; Nerlov, C.; Porse, B.T. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008, 22, 1381–1396. [Google Scholar] [CrossRef]
- Medghalchi, S.M.; Frischmeyer, P.A.; Mendell, J.T.; Kelly, A.G.; Lawler, A.M.; Dietz, H.C. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 2001, 10, 99–105. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Pan, Q.; Reilly, P.T.; Elia, A.J.; McCracken, S.; Wakeham, A.C.; Itie-Youten, A.; Blencowe, B.J.; Mak, T.W. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc. Natl. Acad. Sci. USA 2010, 107, 12186–12191. [Google Scholar] [CrossRef]
- Huang, L.; Shum, E.Y.; Jones, S.H.; Lou, C.-H.; Dumdie, J.; Kim, H.; Roberts, A.J.; Jolly, L.A.; Espinoza, J.L.; Skarbrevik, D.M.; et al. A Upf3b-mutant mouse model with behavioral and neurogenesis defects. Mol. Psychiatry 2018, 23, 1773–1786. [Google Scholar] [CrossRef]
- Futsch, N.; Prates, G.; Mahieux, R.; Casseb, J.; Dutartre, H. Cytokine Networks Dysregulation during HTLV-1 Infection and Associated Diseases. Viruses 2018, 10, 691. [Google Scholar] [CrossRef] [PubMed]
- Kogure, Y.; Kataoka, K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017, 108, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Ware, M.D.; Buisson, M.; Zetoune, A.B.; Stoppa-Lyonnet, D.; Sinilnikova, O.M.; Mazoyer, S. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat. 2008, 29, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Vidoz, L.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Lenoir, G.M.; Mazoyer, S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002, 11, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M.; et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef]
- Karam, R.; Wilkinson, M. A conserved microRNA/NMD regulatory circuit controls gene expression. RNA Biol. 2012, 9, 22–26. [Google Scholar] [CrossRef]
- Gardner, L.B. Nonsense-mediated RNA decay regulation by cellular stress: Implications for tumorigenesis. Mol. Cancer Res. 2010, 8, 295–308. [Google Scholar] [CrossRef]
- Wengrod, J.; Martin, L.; Wang, D.; Frischmeyer-Guerrerio, P.; Dietz, H.C.; Gardner, L.B. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol. Cell. Biol. 2013, 33, 2128–2135. [Google Scholar] [CrossRef]
- Wang, D.; Zavadil, J.; Martin, L.; Parisi, F.; Friedman, E.; Levy, D.; Harding, H.; Ron, D.; Gardner, L.B. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 2011, 31, 3670–3680. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Stoica, L.; Liu, Y.; Zhu, P.J.; Bhattacharya, A.; Buffington, S.A.; Huq, R.; Eissa, N.T.; Larsson, O.; Porse, B.T.; et al. Inhibition of Upf2-Dependent Nonsense-Mediated Decay Leads to Behavioral and Neurophysiological Abnormalities by Activating the Immune Response. Neuron 2019, 104, 665–679.e8. [Google Scholar] [CrossRef] [PubMed]
- Gloggnitzer, J.; Akimcheva, S.; Srinivasan, A.; Kusenda, B.; Riehs, N.; Stampfl, H.; Bautor, J.; Dekrout, B.; Jonak, C.; Jiménez-Gómez, J.M.; et al. Nonsense-Mediated mRNA Decay Modulates Immune Receptor Levels to Regulate Plant Antibacterial Defense. Cell Host Microbe 2014, 16, 376–390. [Google Scholar] [CrossRef]
- Nickless, A.; Jackson, E.; Marasa, J.; Nugent, P.; Mercer, R.W.; Piwnica-Worms, D.; You, Z. Intracellular calcium regulates nonsense-mediated mRNA decay. Nat. Med. 2014, 20, 961–966. [Google Scholar] [CrossRef]
- Ajamian, L.; Abel, K.; Rao, S.; Vyboh, K.; García-de-Gracia, F.; Soto-Rifo, R.; Kulozik, A.E.; Gehring, N.H.; Mouland, A.J. HIV-1 Recruits UPF1 but Excludes UPF2 to Promote Nucleocytoplasmic Export of the Genomic RNA. Biomolecules 2015, 5, 2808–2839. [Google Scholar] [CrossRef]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef]
- Tang, X.; Zhu, Y.; Baker, S.L.; Bowler, M.W.; Chen, B.J.; Chen, C.; Hogg, J.R.; Goff, S.P.; Song, H. Structural basis of suppression of host translation termination by Moloney Murine Leukemia Virus. Nat. Commun. 2016, 7, 12070. [Google Scholar] [CrossRef]
- Kim, Y.K.; Maquat, L.E. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 2019, 25, 407–422. [Google Scholar] [CrossRef]
- Mino, T.; Iwai, N.; Endo, M.; Inoue, K.; Akaki, K.; Hia, F.; Uehata, T.; Emura, T.; Hidaka, K.; Suzuki, Y.; et al. Translation-dependent unwinding of stem-loops by UPF1 licenses Regnase-1 to degrade inflammatory mRNAs. Nucleic Acids Res. 2019, 47, 8838–8859. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Antiviral Factors | Restriction Mechanisms | Potential Involvement in HTLV-1 Restriction | References | |
|---|---|---|---|---|
| TRIM Family | Recognition of the capsid lattice Interference with disassembly of the viral particles. E3 ubiquitin ligase activity targeting viral core component. [19] | TRIM19/PML interferes with HTLV-1 replication via the proteosomal degradation of the viral protein Tax. | + | [20] |
| R136Q TRIM5α variants are correlated with lower proviral loads. | + | [13] | ||
| Negative correlation between HTLV-1 virological parameters or clinical status and the expression of TRIM5α and TRIM22. Negative correlation between Tax mRNA levels and the expression of TRIM19/PML. | + | [14] | ||
| APOBEC Family | C->U editing enzymes leads to stopping codon incorporation after reverse transcription. | APOBEC3G (h3AG) is packaged into HTLV-1 virions. The HTLV-1 genome may be edited in vivo by h3AG, as well as other hA3 members (A, B, C, F, and H). | + | [21,22,23] |
| G-to-A mutations were not detected in the proviruses from infected patients. HTLV-1 Gag protein would reduce the packaging of h3AG into virions. | - | [21,24] | ||
| Loss-of-expression mutations likely generated by hA3G induced mutagenesis in HTLV-1 genes of 60 ATL cases. | + | [25] | ||
| hA3B increased expression in ATL and asymptomatic carriers. | + | [26] | ||
| No correlation with the clinical HAM/TSP states, PVLs or viral mutations. | - | [13] | ||
| Negative correlation between members of APOBEC3 family and Tax mRNA levels. | + | [14] | ||
| In HTLV-1 infected humanized mice showing ATL-like feature, an increased expression level of hA3. No correlation in HAM/TSP cohorts study. | +/- | [27] | ||
| SAMHD1 | SAMHD1 decreases the pool of available dNTPs, inhibiting reverse transcription. Modulates antiviral activity by inhibiting the NF-κB and interferon pathways [28] | No effect on HTLV-1 and Tax expression in macrophages and cycling CD4+ T cells. | - | [15] |
| SAMHD1-mediated apoptotic response in human primary monocytes. | + | [29] | ||
| No correlation with the HAM/TSP parameters. | - | [13] | ||
| SAMHD1 is negatively correlated within HAM/TSP cohorts. | + | [14] | ||
| miRNA | miR-28-3p targets genomic viral mRNA of HTLV-1 strains. | miR-28-3p could inhibit HTLV-1 replication by reducing expression of all viral proteins in a Tax-independent manner. | + | [30] |
| BST2/Tetherin | Inhibits the release of enveloped viruses by tethering them to the cell surface, where they undergo endocytosis and degradation. | No correlation with cell-cell transmission. | - | [31] |
| No correlation with the HAM/TSP parameters. | - | [13] | ||
| BST2 is negatively correlated with HAM/TSP status. | + | [14] | ||
| Zinc finger Antiviral Protein (ZAP) | Targets viral RNA at specific response elements (such as ZRE). Recruits cellular decay factors (PARN, DCP-1, 3‘–5’ exosome). [18] | HTLV-1 is susceptible to a ZAP-mediated viral RNA processing during early infection. | + | [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prochasson, L.; Jalinot, P.; Mocquet, V. The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD). Pathogens 2020, 9, 287. https://doi.org/10.3390/pathogens9040287
Prochasson L, Jalinot P, Mocquet V. The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD). Pathogens. 2020; 9(4):287. https://doi.org/10.3390/pathogens9040287
Chicago/Turabian StyleProchasson, Léa, Pierre Jalinot, and Vincent Mocquet. 2020. "The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD)" Pathogens 9, no. 4: 287. https://doi.org/10.3390/pathogens9040287
APA StyleProchasson, L., Jalinot, P., & Mocquet, V. (2020). The Complex Relationship between HTLV-1 and Nonsense-Mediated mRNA Decay (NMD). Pathogens, 9(4), 287. https://doi.org/10.3390/pathogens9040287