The 3′ Untranslated Region of a Plant Viral RNA Directs Efficient Cap-Independent Translation in Plant and Mammalian Systems





Abstract
1. Introduction
2. Results
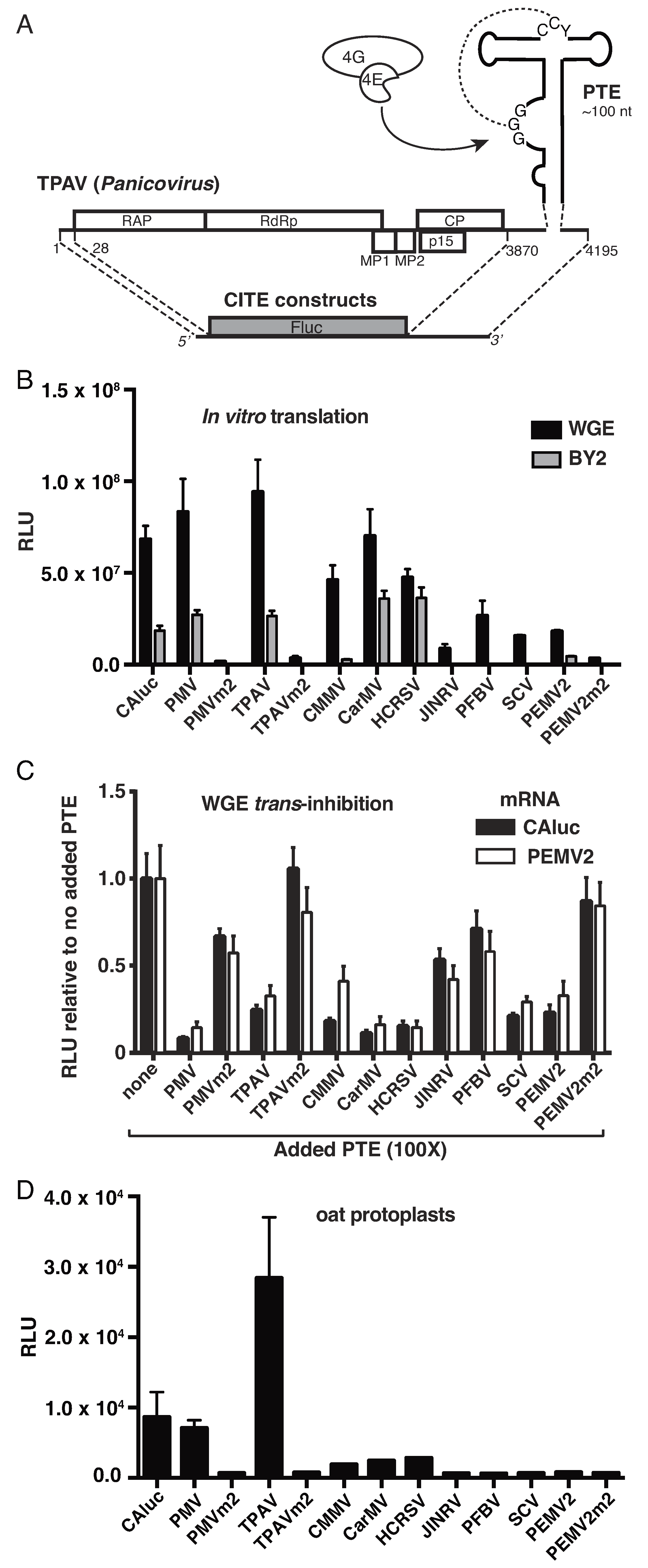
2.1. Thin Paspalum Asymptomatic Virus (TPAV) Has the Most Powerful PTE
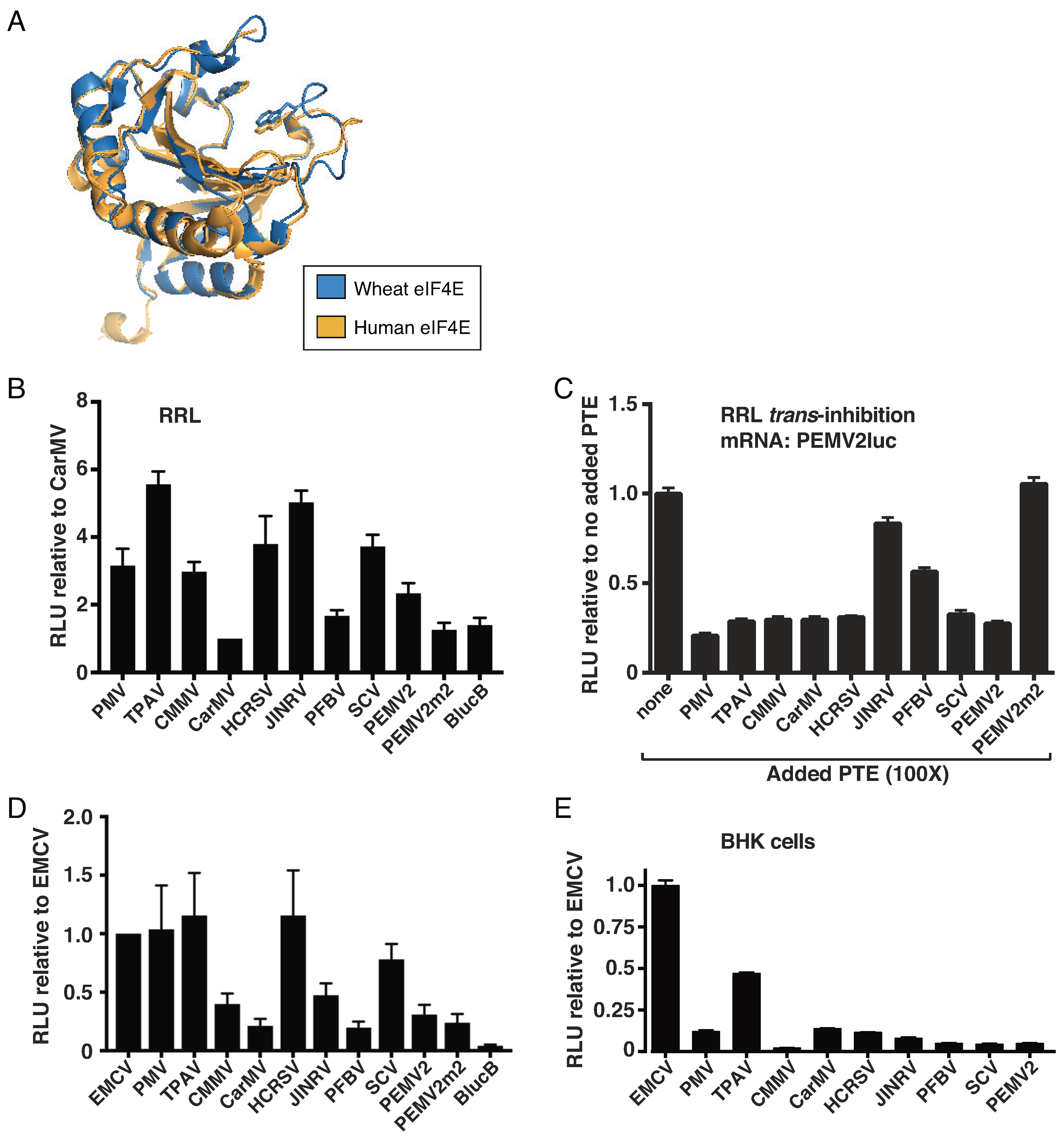
2.2. The TPAV PTE Confers Efficient Cap-Independent Translation in Mammalian Extracts and Cells
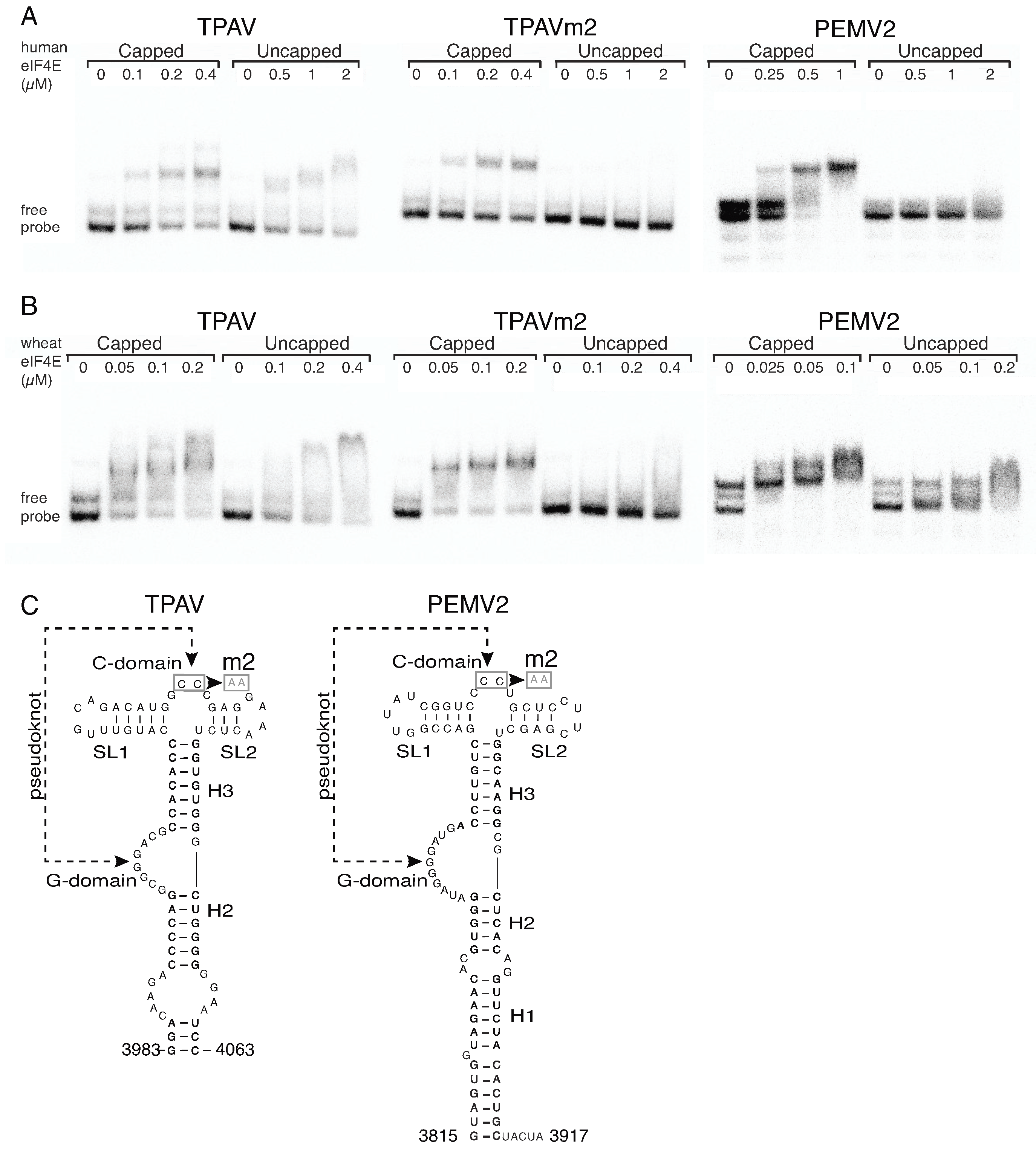
2.3. The TPAV PTE Binds Human eIF4E
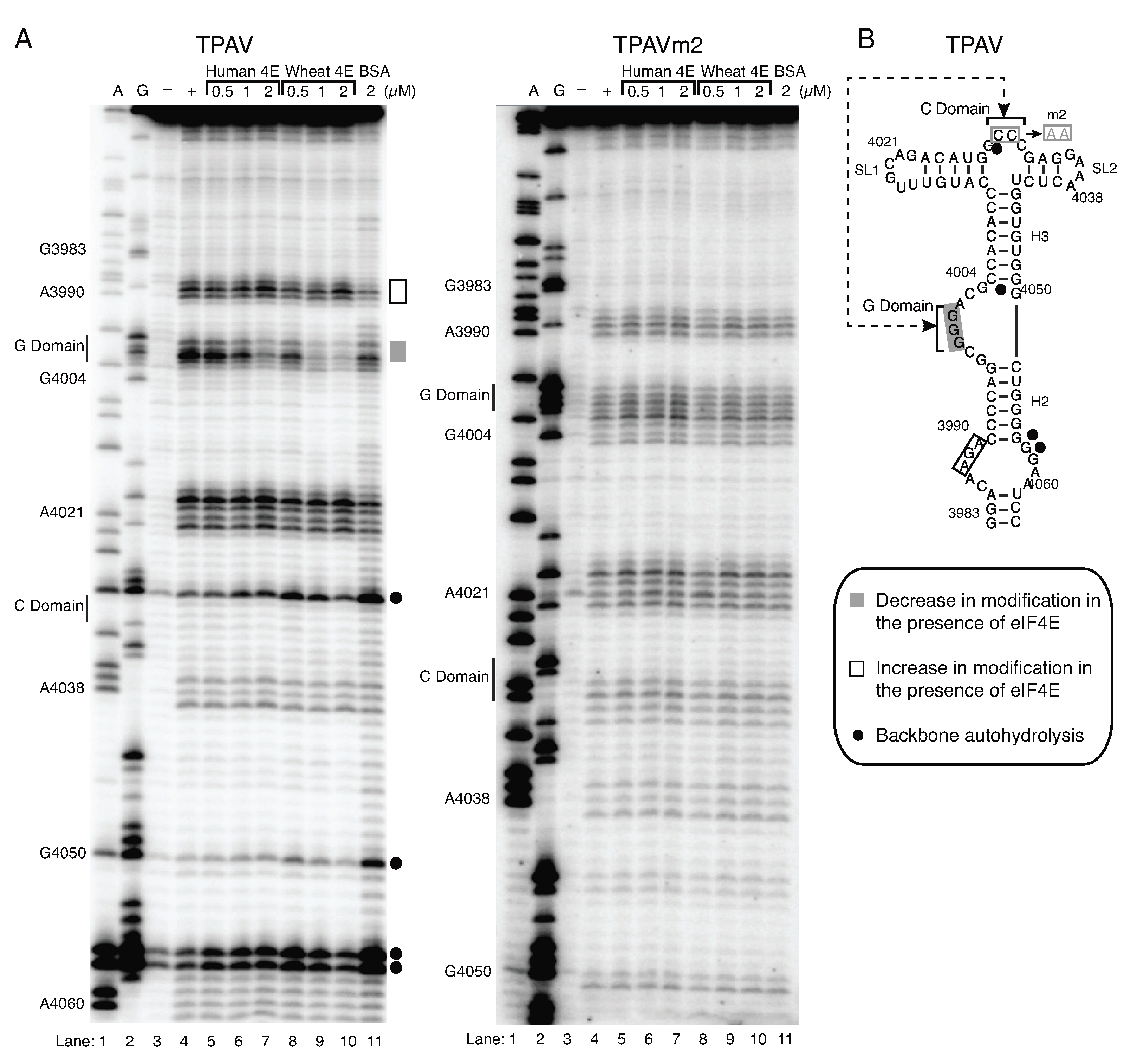
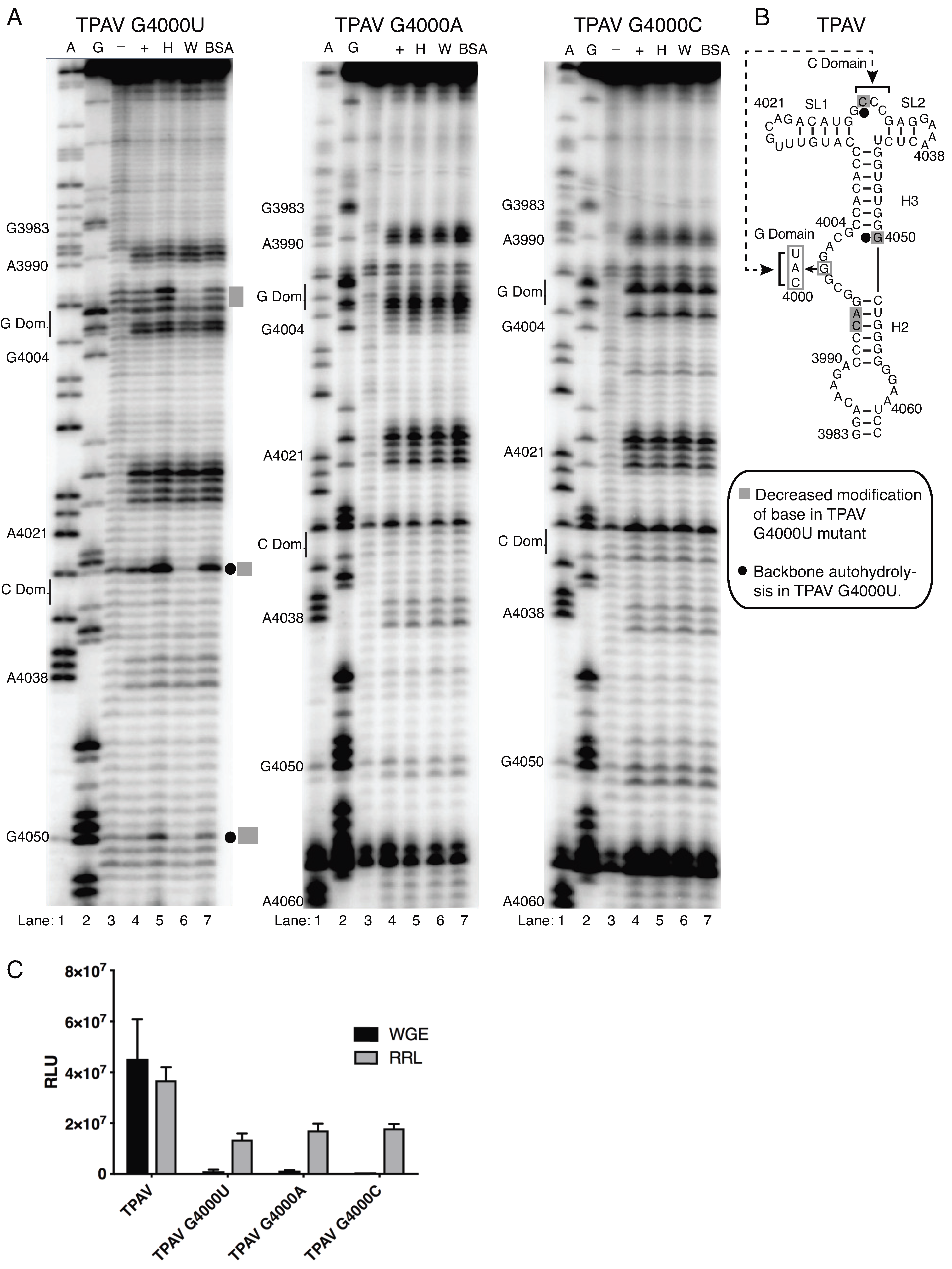
2.4. The G-Rich Bulge (G-Domain) of the PTE Is Involved in eIF4E Binding
3. Discussion
3.1. Interaction of Mammalian eIF4E with TPAV PTE
3.2. Why No (known) 3′ CITEs in Animals?
4. Materials and Methods
4.1. eIF4E Structure Alignment
4.2. Plasmid Constructs and RNA Synthesis
4.3. Recombinant Protein Expression
4.4. Translation in Plant Systems
4.5. Translation in the Mammalian Systems
4.6. Electrophoretic Mobility Shift Assay (EMSA)
4.7. RNA Structure Probing and Footprinting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flint, S.J.; Racaniello, V.R.; Rall, G.F.; Skalka, A.M.; Enquist, L.W. Principles of Virology, 3rd ed.; ASM Press: Washington, DC, USA, 2015. [Google Scholar]
- Ahola, T.; Ahlquist, P. Putative RNA capping activities encoded by brome mosaic virus: Methylation and covalent binding of guanylate by replicase protein 1a. J. Virol. 1999, 73, 10061–10069. [Google Scholar] [PubMed]
- Leen, E.N.; Sorgeloos, F.; Correia, S.; Chaudhry, Y.; Cannac, F.; Pastore, C.; Xu, Y.; Graham, S.C.; Matthews, S.J.; Goodfellow, I.G.; et al. A Conserved Interaction between a C-Terminal Motif in Norovirus VPg and the HEAT-1 Domain of eIF4G Is Essential for Translation Initiation. PLoS Pathog. 2016, 12, e1005379. [Google Scholar]
- Sweeney, T.R.; Abaeva, I.S.; Pestova, T.V.; Hellen, C.U. The mechanism of translation initiation on Type 1 picornavirus IRESs. EMBO J. 2014, 33, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Unbehaun, A.; Spahn, C.M.T. Ribosomal Chamber Music: Toward an Understanding of IRES Mechanisms. Trends Biochem. Sci. 2017, 42, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Mailliot, J.; Martin, F. Viral internal ribosomal entry sites: Four classes for one goal. Wiley Interdiscip. Rev. RNA 2018, 9, e1458. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Zhang, J.; Mayberry, L.K.; Tatineni, S.; Browning, K.S.; Rakotondrafara, A.M. A Unique 5′ Translation Element Discovered in Triticum Mosaic Virus. J. Virol. 2015, 89, 12427–12440. [Google Scholar] [CrossRef] [PubMed]
- Zeenko, V.; Gallie, D.R. Cap-independent translation of tobacco etch virus is conferred by an RNA pseudoknot in the 5′-leader. J. Biol. Chem. 2005, 280, 26813–26824. [Google Scholar] [CrossRef] [PubMed]
- Miras, M.; Miller, W.A.; Truniger, V.; Aranda, M.A. Non-canonical Translation in Plant RNA Viruses. Front. Plant Sci. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Roberts, R.; Rakotondrafara, A.M. The role of the 5′ untranslated regions of Potyviridae in translation. Virus Res. 2015, 206, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Karetnikov, A.; Lehto, K. Translation mechanisms involving long-distance base pairing interactions between the 5′ and 3′ non-translated regions and internal ribosomal entry are conserved for both genomic RNAs of Blackcurrant reversion nepovirus. Virology 2008, 371, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Allen, E.; Miller, W.A. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol. Cell 2001, 7, 1103–1109. [Google Scholar] [CrossRef]
- Nicholson, B.L.; White, K.A. 3′ cap-independent translation enhancers of positive-strand RNA plant viruses. Curr. Opin. Virol. 2011, 1, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.E.; Miller, W.A. 3′ cap-independent translation enhancers of plant viruses. Annu. Rev. Microbiol. 2013, 67, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Treder, K.; Miller, W.A. Structure of a viral cap-independent translation element that functions via high affinity binding to the eIF4E subunit of eIF4F. J. Biol. Chem. 2009, 284, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Treder, K.; Pettit Kneller, E.L.; Allen, E.M.; Wang, Z.; Browning, K.S.; Miller, W.A. The 3′ cap-independent translation element of Barley yellow dwarf virus binds eIF4F via the eIF4G subunit to initiate translation. RNA 2008, 14, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.L.; Zaslaver, O.; Mayberry, L.K.; Browning, K.S.; White, K.A. Tombusvirus Y-shaped translational enhancer forms a complex with eIF4F and can be functionally replaced by heterologous translational enhancers. J. Virol. 2013, 87, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.L.; Wu, B.; Chevtchenko, I.; White, K.A. Tombusvirus recruitment of host translational machinery via the 3′ UTR. RNA 2010, 16, 1402–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, Q.; Miller, W.A.; Goss, D.J. Eukaryotic translation initiation factor 4G (eIF4G) coordinates interactions with eIF4A, eIF4B and eIF4E in binding and translation of the barley yellow dwarf virus 3′ cap-independent translation element (BTE). J. Biol. Chem. 2017, 292, 5921–5931. [Google Scholar] [CrossRef] [PubMed]
- Miras, M.; Rodriguez-Hernandez, A.M.; Romero-Lopez, C.; Berzal-Herranz, A.; Colchero, J.; Aranda, M.A.; Truniger, V. A Dual Interaction between the 5′- and 3′-ends of the melon necrotic spot virus (MNSV) RNA genome is required for efficient cap-independent translation. Front. Plant Sci. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Stupina, V.A.; Yuan, X.; Meskauskas, A.; Dinman, J.D.; Simon, A.E. Ribosome binding to a 5′ translational enhancer is altered in the presence of the 3′ untranslated region in cap-independent translation of turnip crinkle virus. J. Virol. 2011, 85, 4638–4653. [Google Scholar] [CrossRef] [PubMed]
- Timmer, R.T.; Benkowski, L.A.; Schodin, D.; Lax, S.R.; Metz, A.M.; Ravel, J.M.; Browning, K.S. The 5′ and 3′ untranslated regions of satellite tobacco necrosis virus RNA affect translational efficiency and dependence on a 5′ cap structure. J. Biol. Chem. 1993, 268, 9504–9510. [Google Scholar] [PubMed]
- van Lipzig, R.; Gultyaev, A.P.; Pleij, C.W.; van Montagu, M.; Cornelissen, M.; Meulewaeter, F. The 5′ and 3′ extremities of the satellite tobacco necrosis virus translational enhancer domain contribute differentially to stimulation of translation. RNA 2002, 8, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Allen, E.; Miller, W.A. Structure and function of a cap-independent translation element that functions in either the 3′ or the 5′ untranslated region. RNA 2000, 6, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, H.; Tatsuta, M.; Kaido, M.; Mise, K.; Okuno, T. Cap-independent translational enhancement by the 3′ untranslated region of red clover necrotic mosaic virus RNA1. J. Virol. 2003, 77, 12113–12121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kraft, J.J.; Hui, A.Y.; Miller, W.A. Structural plasticity of Barley yellow dwarf virus-like cap-independent translation elements in four genera of plant viral RNAs. Virology 2010, 402, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Truniger, V.; Nieto, C.; Gonzalez-Ibeas, D.; Aranda, M. Mechanism of plant eIF4E-mediated resistance against a Carmovirus (Tombusviridae): Cap-independent translation of a viral RNA controlled in cis by an (a)virulence determinant. Plant J. 2008, 56, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Wang, J.; Yu, P.; Eyler, D.; Xu, H.; Starich, M.R.; Tiede, D.M.; Simon, A.E.; Kasprzak, W.; Schwieters, C.D.; et al. Solution structure of the cap-independent translational enhancer and ribosome-binding element in the 3′ UTR of turnip crinkle virus. Proc. Natl. Acad. Sci. USA 2010, 107, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Kasprzak, W.K.; Kim, T.; Gao, F.; Young, M.Y.; Yuan, X.; Shapiro, B.A.; Seog, J.; Simon, A.E. Folding behavior of a T-shaped, ribosome-binding translation enhancer implicated in a wide-spread conformational switch. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Stupina, V.A.; Meskauskas, A.; McCormack, J.C.; Yingling, Y.G.; Shapiro, B.A.; Dinman, J.D.; Simon, A.E. The 3′ proximal translational enhancer of Turnip crinkle virus binds to 60S ribosomal subunits. RNA 2008, 14, 2379–2393. [Google Scholar] [CrossRef] [PubMed]
- Batten, J.S.; Desvoyes, B.; Yamamura, Y.; Scholthof, K.B. A translational enhancer element on the 3′-proximal end of the Panicum mosaic virus genome. FEBS Lett. 2006, 580, 2591–2597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Parisien, M.; Scheets, K.; Miller, W.A. The cap-binding translation initiation factor, eIF4E, binds a pseudoknot in a viral cap-independent translation element. Structure 2011, 19, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Miras, M.; Sempere, R.N.; Kraft, J.J.; Miller, W.A.; Aranda, M.A.; Truniger, V. Interfamilial recombination between viruses led to acquisition of a novel translation-enhancing RNA element that allows resistance breaking. New Phytol. 2014, 202, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Hellen, C.U.; Pestova, T.V. Toward the mechanism of eIF4F-mediated ribosomal attachment to mammalian capped mRNAs. Genes Dev. 2016, 30, 1573–1588. [Google Scholar] [CrossRef] [PubMed]
- Gazo, B.M.; Murphy, P.; Gatchel, J.R.; Browning, K.S. A novel interaction of Cap-binding protein complexes eukaryotic initiation factor (eIF) 4F and eIF(iso)4F with a region in the 3′-untranslated region of satellite tobacco necrosis virus. J. Biol. Chem. 2004, 279, 13584–13592. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Shi, K.; Meskauskas, A.; Simon, A.E. The 3′ end of Turnip crinkle virus contains a highly interactive structure including a translational enhancer that is disrupted by binding to the RNA-dependent RNA polymerase. RNA 2009, 15, 1849–1864. [Google Scholar] [CrossRef] [PubMed]
- Miras, M.; Truniger, V.; Querol-Audi, J.; Aranda, M.A. Analysis of the interacting partners eIF4F and 3′-CITE required for Melon necrotic spot virus cap-independent translation. Mol. Plant Pathol. 2017, 18, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.-C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Browning, K.S.; Webster, C.; Roberts, J.K.; Ravel, J.M. Identification of an isozyme form of protein synthesis initiation factor 4F in plants. J. Biol. Chem. 1992, 267, 10096–10100. [Google Scholar] [PubMed]
- Patrick, R.M.; Browning, K.S. The eIF4F and eIFiso4F Complexes of Plants: An Evolutionary Perspective. Comp. Funct. Genom. 2012, 2012, 287814. [Google Scholar] [CrossRef] [PubMed]
- Scheets, K. Infectious transcripts of an asymptomatic panicovirus identified from a metagenomic survey. Virus Res. 2013, 176, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.J.; Treder, K.; Peterson, M.S.; Miller, W.A. Cation-dependent folding of 3′ cap-independent translation elements facilitates interaction of a 17-nucleotide conserved sequence with eIF4G. Nucleic Acids Res. 2013, 41, 3398–3413. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Alekhina, O.M.; Vassilenko, K.S.; Simon, A.E. Concerted action of two 3′ cap-independent translation enhancers increases the competitive strength of translated viral genomes. Nucleic Acids Res. 2017, 45, 9558–9572. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, F.; Simon, A.E. Differential use of 3′ CITEs by the subgenomic RNA of Pea enation mosaic virus 2. Virology 2017, 510, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Kasprzak, W.K.; Szarko, C.; Shapiro, B.A.; Simon, A.E. The 3′ Untranslated Region of Pea Enation Mosaic Virus Contains Two T-Shaped, Ribosome-Binding, Cap-Independent Translation Enhancers. J. Virol. 2014, 88, 11696–11712. [Google Scholar] [CrossRef] [PubMed]
- Carrington, J.C.; Morris, T.J. Characterization of the cell-free translation products of carnation mottle virus genomic and subgenomic RNAs. Virology 1985, 144, 1–10. [Google Scholar] [CrossRef]
- Fan, Q.; Treder, K.; Miller, W.A. Untranslated regions of diverse plant viral RNAs vary greatly in translation enhancement efficiency. BMC Biotechnol. 2012, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Miller, W.A. A sequence located 4.5 to 5 kilobases from the 5′ end of the barley yellow dwarf virus (PAV) genome strongly stimulates translation of uncapped mRNA. J. Biol. Chem. 1995, 270, 13446–13452. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Palmenberg, A.C. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques 2006, 41, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Kumar, P.; Hellen, C.U.; D’Souza, V.M.; Wagner, G. An accurately preorganized IRES RNA structure enables eIF4G capture for initiation of viral translation. Nat. Struct. Mol. Biol. 2016, 23, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Rifo, R.S.; Ricci, E.P.; Décimo, D.; Moncorgé, O.; Ohlmann, T. Back to basics: the untreated rabbit reticulocyte lysate as a competitive system to recapitulate cap/poly(A) synergy and the selective advantage of IRES-driven translation. Nucleic Acids Res. 2007, 35, e121. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [PubMed]
- Koh, D.C.; Liu, D.X.; Wong, S.M. A six-nucleotide segment within the 3′ untranslated region of hibiscus chlorotic ringspot virus plays an essential role in translational enhancement. J. Virol. 2002, 76, 1144–1153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Svitkin, Y.V.; Ovchinnikov, L.P.; Dreyfuss, G.; Sonenberg, N. General RNA binding proteins render translation cap dependent. EMBO J. 1996, 15, 7147–7155. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Kasprzak, W.; Stupina, V.A.; Shapiro, B.A.; Simon, A.E. A ribosome-binding, 3′ translational enhancer has a T-shaped structure and engages in a long-distance RNA-RNA interaction. J. Virol. 2012, 86, 9828–9842. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Mesa, H.; Gannon, M.; Holshbach, E.; Zhang, J.; Roberts, R.; Buettner, M.; Rakotondrafara, A.M. Triticum mosaic virus IRES relies on a picornavirus-like YX-AUG motif to designate the preferred translation initiation site and to likely target the 18S rRNA. J. Virol. 2019, 93, e01705-18. [Google Scholar] [PubMed]
- Bai, Y.; Zhou, K.; Doudna, J.A. Hepatitis C virus 3′ UTR regulates viral translation through direct interactions with the host translation machinery. Nucleic Acids Res. 2013, 41, 7861–7874. [Google Scholar] [CrossRef] [PubMed]
- Bung, C.; Bochkaeva, Z.; Terenin, I.; Zinovkin, R.; Shatsky, I.N.; Niepmann, M. Influence of the hepatitis C virus 3′-untranslated region on IRES-dependent and cap-dependent translation initiation. FEBS Lett. 2010, 584, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M.; Shalamova, L.A.; Gerresheim, G.K.; Rossbach, O. Signals Involved in Regulation of Hepatitis C Virus RNA Genome Translation and Replication. Front. Microbiol. 2018, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Sanz, M.A.; Carrasco, L. A Viral mRNA Motif at the 3′-Untranslated Region that Confers Translatability in a Cell-Specific Manner. Implications for Virus Evolution. Sci. Rep. 2016, 6, 19217. [Google Scholar] [CrossRef] [PubMed]
- Holden, K.L.; Harris, E. Enhancement of dengue virus translation: Role of the 3′ untranslated region and the terminal 3′ stem-loop domain. Virology 2004, 329, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Reichert, E.D.; Polo, S.; Falgout, B.; Kasprzak, W.; Shapiro, B.A.; Padmanabhan, R. Identification of cis-acting elements in the 3′-untranslated region of the dengue virus type 2 RNA that modulate translation and replication. J. Biol. Chem. 2011, 286, 22521–22534. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Qin, C.; Jiang, T.; Li, X.; Zhao, H.; Liu, Z.; Deng, Y.; Liu, R.; Chen, S.; Yu, M.; et al. Translational regulation by the 3′ untranslated region of the dengue type 2 virus genome. Am. J. Trop. Med. Hyg. 2009, 81, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kastan, M.B. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev. 2010, 24, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Barends, S.; Jaeger, S.; Schaeffer, L.; Prongidi-Fix, L.; Eriani, G. Cap-assisted internal initiation of translation of histone H4. Mol. Cell 2011, 41, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.O.; Mizumoto, H.; Nagano, H.; Imoto, Y.; Takigawa, K.; Sarawaneeyaruk, S.; Kaido, M.; Mise, K.; Okuno, T. A viral noncoding RNA generated by cis-element-mediated protection against 5′->3′ RNA decay represses both cap-independent and cap-dependent translation. J. Virol. 2008, 82, 10162–10174. [Google Scholar] [CrossRef] [PubMed]
- Steckelberg, A.-L.; Vicens, Q.; Kieft, J.S. Exoribonuclease-Resistant RNAs Exist within both Coding and Noncoding Subgenomic RNAs. mBio 2018, 9, e02461-18. [Google Scholar] [CrossRef] [PubMed]
- Dilweg, I.W.; Gultyaev, A.P.; Olsthoorn, R. A widespread Xrn1-resistant RNA motif composed of two short hairpins. bioRxiv 2019, 522318. [Google Scholar]
- Barry, J.K.; Miller, W.A. A-1 ribosomal frameshift element that requires base pairing across four kilobases suggests a mechanism of regulating ribosome and replicase traffic on a viral RNA. Proc. Natl. Acad. Sci. USA 2002, 99, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.A.; White, K.A. Long distance RNA-RNA interactions in plant virus gene expression and replication. Ann. Rev. Phytopathol. 2006, 44, 447–467. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Najita, L.; Franzusoff, A.; Sarnow, P. Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol. Cell Biol. 1994, 14, 7322–7330. [Google Scholar] [CrossRef] [PubMed]
- Komoda, K.; Naito, S.; Ishikawa, M. Replication of plant RNA virus genomes in a cell-free extract of evacuolated plant protoplasts. Proc. Natl. Acad. Sci. USA 2004, 101, 1863–1867. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Miller, W.A. The 3′ untranslated region of tobacco necrosis virus RNA contains a barley yellow dwarf virus-like cap-independent translation element. J. Virol. 2004, 78, 4655–4664. [Google Scholar] [CrossRef] [PubMed]
- Rakotondrafara, A.M.; Jackson, J.; Pettit Kneller, E.J.; Miller, W.A. Preparation and electroporation of oat protoplasts from cell suspension culture. In Current Protocols in Microbiology; Coico, R., Towalik, T., Quaries, J., Stevenson, B., Taylor, R., Eds.; John Wiley & Sons: New York, NY, USA, 2007; pp. 16D.3.1–16D.3.12. [Google Scholar]
- Habjan, M.; Penski, N.; Spiegel, M.; Weber, F. T7 RNA polymerase-dependent and -independent systems for cDNA-based rescue of Rift Valley fever virus. J. Gen. Virol. 2008, 89, 2157–2166. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kd (nM) | ||||||
| TPAV PTE | TPAV m2 PTE | PEMV RNA2 PTE | ||||
| eIF4Es | capped | uncapped | capped | uncapped | capped | uncapped |
| human | 160 ± 1 | 838 ± 124 | 253 ± 45 | 6362 ± 1283 | 285 ± 33 | 2524 ± 435 |
| wheat | 31 ± 7 | 65 ± 14 | 26 ± 2 | 463 ± 185 | 41 ± 25 | 110 ± 38 |
| Fold Kd human/wheat eIF4E | ||||||
| TPAV PTE | TPAV m2 PTE | PEMV RNA2 PTE | ||||
| capped | uncapped | capped | uncapped | capped | uncapped | |
| 5.16 *** | 12.89 ** | 9.73 ** | 13.74 ** | 6.95 *** | 22.94 ** | |
| Fold Kd uncapped/capped RNA | ||||||
| TPAV PTE | TPAV m2 PTE | PEMV RNA2 PTE | ||||
| human | wheat | human | wheat | human | wheat | |
| 5.24 ** | 2.10 * | 25.15 ** | 17.81 * | 8.86 ** | 2.68 * | |
| Fold Kd TPAVm2/TPAV RNA | ||||||
| human eIF4E | wheat eIF4E | |||||
| capped | uncapped | capped | uncapped | |||
| 1.58 * | 7.59 ** | 0.84 n.s | 7.12 * | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, J.J.; Peterson, M.S.; Cho, S.K.; Wang, Z.; Hui, A.; Rakotondrafara, A.M.; Treder, K.; Miller, C.L.; Miller, W.A. The 3′ Untranslated Region of a Plant Viral RNA Directs Efficient Cap-Independent Translation in Plant and Mammalian Systems. Pathogens 2019, 8, 28. https://doi.org/10.3390/pathogens8010028
Kraft JJ, Peterson MS, Cho SK, Wang Z, Hui A, Rakotondrafara AM, Treder K, Miller CL, Miller WA. The 3′ Untranslated Region of a Plant Viral RNA Directs Efficient Cap-Independent Translation in Plant and Mammalian Systems. Pathogens. 2019; 8(1):28. https://doi.org/10.3390/pathogens8010028
Chicago/Turabian StyleKraft, Jelena J., Mariko S. Peterson, Sung Ki Cho, Zhaohui Wang, Alice Hui, Aurélie M. Rakotondrafara, Krzysztof Treder, Cathy L. Miller, and W. Allen Miller. 2019. "The 3′ Untranslated Region of a Plant Viral RNA Directs Efficient Cap-Independent Translation in Plant and Mammalian Systems" Pathogens 8, no. 1: 28. https://doi.org/10.3390/pathogens8010028
APA StyleKraft, J. J., Peterson, M. S., Cho, S. K., Wang, Z., Hui, A., Rakotondrafara, A. M., Treder, K., Miller, C. L., & Miller, W. A. (2019). The 3′ Untranslated Region of a Plant Viral RNA Directs Efficient Cap-Independent Translation in Plant and Mammalian Systems. Pathogens, 8(1), 28. https://doi.org/10.3390/pathogens8010028