

Novel Thiazolylimidazole Hybrids as Promising Antileishmanial Agents: Rational Design and Biological Evaluation


Abstract
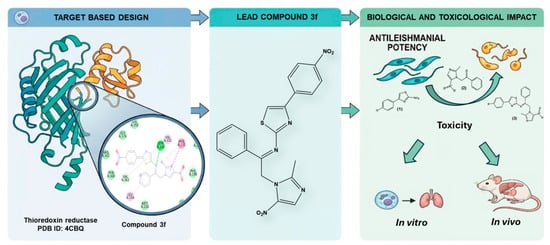
1. Introduction
2. Materials and Methods
2.1. In Silico Affinity Mapping Procedure
2.2. General Procedure for Synthesis of 2-Amino-4-arylthiazoles (1)
2.2.1. Data of 2-amino-4-phenyl Thiazole (1a)
2.2.2. Data of 2-amino-4-p-tolyl Thiazole (1b)
2.2.3. Data of 2-amino-4-fluorophenyl Thiazole (1c)
2.2.4. Data of 2-amino-4-chlorophenyl Thiazole (1d)
2.2.5. Data of 2-amino-4-cyanophenyl Thiazole (1e)
2.2.6. Data of 2-amino-4-nitrophenyl Thiazole (1f)
2.2.7. Data of 3-(N)-phenacyl-2-methyl-5-nitroimidazole (2)
2.3. General Procedure for Synthesis of the 1-(2-Methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-phenylthiazol-2-yl)ethan-1-imine (Series 3)
2.3.1. Data of 1-(2-methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-phenylthiazol-2-yl)ethan-1-imine (3a)
2.3.2. Data of 1-(2-methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-(p-tolyl)thiazol-2-yl)ethan-1-imine (3b)
2.3.3. Data of 1-(2-methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-(p-fluoro)thiazol-2-yl)ethan-1-imine (3c)
2.3.4. Data of 1-(2-methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-(p-chloro)thiazol-2-yl)ethan-1-imine (3d)
2.3.5. Data of 1-(2-methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-(p-nitro)thiazol-2-yl)ethan-1-imine (3f)
2.4. Biological Evaluation
2.4.1. In Vitro Biological Assays
Determination of Leishmanicidal Activity
Cytotoxicity Assays in Vero Cells
2.4.2. In Vivo Evaluation of Acute Toxicity
Animals Care and Housing
Determination of Medium Letal Dose (LD50)
2.5. Statistical Analysis
3. Results
3.1. In Silico Analysis
3.2. Synthesis of Antileishmanial Compounds
3.2.1. Synthesis of 2-Amino-4-phenyl thiazoles [34]
3.2.2. Synthesis of 1-(2-Methyl-5-nitro-1H-imidazol-1-yl)-2-phenyl-N-(4-R-phenylthiazol-2-yl) ethan-1-imine (Series 3)
3.3. In Vitro Studies
Antileishmanial Response and Cytotoxicity
3.4. In Vivo Studies (Acute Toxicity Study)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American Type Culture Collection |
| ATR | Attenuated Total Reflectance |
| b.w. | Body Weight |
| CC50 | Cytotoxic concentration 50% |
| CEI | Ethics and Research Committee |
| CH3 | Methyl group |
| Cl | Chloro group |
| CL | Cutaneous Leishmaniasis |
| CN | Cyano group |
| COSY | Correlation System Spectroscopy |
| DMSO | Dimethyl Sulfoxide |
| F | Fluoro group |
| FBS | Fetal Bovine Serum |
| ∆G | Gibbs Free Energy (binding free energy) |
| H | Hydrogen |
| HMBC | Heteronuclear Multiple Bond Correlation |
| IC50 | Inhibitory concentration 50% |
| IC90 | Inhibitory concentration 90% |
| IR | Infrared Spectra |
| Ka/∆G | Binding energy |
| KI | Inhibitory Constant |
| LGA | Lamarckian Genetic Algorithm |
| LD | Lethal dose |
| LD50 | Lethal dose 50% |
| LogP | Partition coefficient |
| MIC | Minimum inhibitory concentration |
| MTF | Miltefosine |
| NO2 | Nitro group |
| OECD | Organization for Economic Co-operation and Development (Test 423) |
| PDB | Protein Data Bank |
| Ph | Phenyl group |
| PROBIT | Probability Unit Regression Method |
| RPMI | Roswell Park Memorial Institute Medium |
| TLC | Thin Layer Chromatography |
| TR | Thioredoxin reductase |
| TryR | Trypanothione reductase |
References
- World Health Organization. Global Report on Neglected Tropical Diseases 2025; WHO: Geneva, Switzerland, 2025. [Google Scholar]
- World Health Organization. Ending the Neglect to Attain the SDGs: A Road Map for Neglected Tropical Diseases 2021–2030; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- World Health Organization. Leishmaniasis: Key Facts; WHO: Geneva, Switzerland, 2024. [Google Scholar]
- Kaye, P.M.; Cruz, I.; Picado, A.; Van Bocxlaer, K.; Croft, S.L. Leishmaniasis immunopathology: Impact on design and use of vaccines, diagnostics and drugs. Semin. Immunopathol. 2020, 42, 247–264. [Google Scholar] [CrossRef]
- Kumari, D.; Perveen, S.; Sharma, R.; Singh, K. Advancement in leishmaniasis diagnosis and therapeutics: An update. Eur. J. Pharmacol. 2021, 910, 174436. [Google Scholar] [CrossRef]
- Van Dijk, N.J.; Carter, J.; Kiptanui, D.; Mens, P.F.; Schallig, H.D.F.H. A case-control study on risk factors for visceral leishmaniasis in West Pokot County, Kenya. Trop. Med. Int. Health 2024, 29, 904–912. [Google Scholar] [CrossRef]
- Sundar, S.; Sinha, P.R.; Agrawal, N.K.; Srivastava, R.; Rainey, P.M.; Berman, J.D.; Murray, H.W.; Singh, V.P. A cluster of cases of severe cardiotoxicity among kala-azar patients treated with a high-osmolarity lot of sodium antimony gluconate. Am. J. Trop. Med. Hyg. 1998, 59, 139–143. [Google Scholar] [CrossRef]
- Gasser, R.A.; Magill, A.J.; Oster, C.N.; Franke, E.D.; Grögl, M.; Berman, J.D. Pancreatitis from pentavalent antimonials. Clin. Infect. Dis. 1994, 18, 83–90. [Google Scholar] [CrossRef]
- Thakur, C.P.; Sinha, G.P.; Pandey, A.K.; Kumar, N.; Kumar, P.; Hassan, S.M.; Narain, S.; Roy, R.K. Efficacy decline of stibogluconate. Ann. Trop. Med. Parasitol. 1998, 92, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Yardley, V. Chemotherapy of leishmaniasis. Curr. Pharm. Des. 2002, 8, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Hafiz, S.; Kyriakopoulos, C. Pentamidine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557586/ (accessed on 23 March 2026).
- Pinto-Martinez, A.K.; Rodriguez-Durán, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of miltefosine. Antimicrob. Agents Chemother. 2017, 62, e01614-17. [Google Scholar] [CrossRef] [PubMed]
- Espada, C.R.; Magalhães, R.M.; Cruz, M.C.; Machado, P.R.; Schriefer, A.; Carvalho, E.M.; Hornillos, V.; Alves, J.M.; Cruz, A.K.; Coelho, A.C.; et al. Investigation of the pathways related to intrinsic miltefosine tolerance in Leishmania (Viannia) braziliensis clinical isolates reveals differences in drug uptake. Int. J. Parasitol. Drugs. Drug. Resist. 2019, 11, 139–147. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Parsonage, D.; Sheng, F.; Hirata, K.; Debnath, A.; McKerrow, J.H.; Reed, S.L.; Abagyan, R.; Poole, L.B.; Podust, L.M. X-ray structures of thioredoxin and thioredoxin reductase from Entamoeba histolytica and prevailing hypothesis of the mechanism of Auranofin action. J. Struct. Biol. 2016, 194, 180–190. [Google Scholar] [CrossRef]
- Chhabria, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef]
- Alghamdi, S.S.; Suliman, R.S.; Almutairi, K.; Kahtani, K.; Aljatli, D. Imidazole as a Promising Medicinal Scaffold: Current Status and Future Direction. Drug Des. Devel. Ther. 2021, 15, 3289–3312. [Google Scholar] [CrossRef]
- Aldrete, M.E.C.; Salgado-Zamora, H.; Luna, J.; Meléndez, E.; Meráz-Ríos, M.A. Nitroimidazoles anti-trichomonas. Toxicol. In Vitro 2005, 8, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Pérez, M. Anti-inflammatory thiazoles. Eur. J. Med. Chem. 2019, 180, 250–258. [Google Scholar]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. [Google Scholar] [CrossRef]
- Bates, P.A. Complete developmental cycle of Leishmania mexicana in vitro. Parasitology 1994, 108, 1. [Google Scholar] [CrossRef] [PubMed]
- Åsberg, A.; Johnsen, H.; Mikkelsen, G.; Hov, G.G. Using probit regression to disclose the analytical performance of qualitative and semi-quantitative tests. Scand. J. Clin. Lab Investig. 2016, 76, 515–519. [Google Scholar] [CrossRef]
- SAGARPA (México). NOM-062-ZOO-1999: Technical Specifications for the Production, Care and Use of Laboratory Animals. Official Journal of the Federation (DOF), Mexico, 22 August 2001.
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- OECD. Test No. 423: Acute Oral Toxicity; OECD Publishing: Paris, France, 2002. [Google Scholar] [CrossRef]
- Gómez-Arroyo, S.; Melchor-Castro, S.; Villalobos-Pietrini, R.; Camargo, E.M.; Salgado-Zamora, H.; Campos Aldrete, M.E. Cytogenetic study of metronidazole and three metronidazole analogues in cultured human lymphocytes with and without metabolic activation. Toxicol. In Vitro 2004, 18, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.S.; Zhang, Y.; Berriman, M.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. Crystal structure of Trypanosoma cruzi trypanothione reductase in complex with trypanothione, and the structure-based discovery of new natural product inhibitors. Structure 1999, 7, 81–89. [Google Scholar] [CrossRef]
- Müller, S.; Liebau, E.; Walter, R.D.; Krauth-Siegel, R.L. Thiol-based redox metabolism of protozoan parasites. Trends Parasitol. 2003, 19, 320–328. [Google Scholar] [CrossRef] [PubMed]
- González-Montero, M.-C.; Andrés-Rodríguez, J.; García-Fernández, N.; Pérez-Pertejo, Y.; Reguera, R.M.; Balaña-Fouce, R.; García-Estrada, C. Targeting Trypanothione Metabolism in Trypanosomatids. Molecules 2024, 29, 2214. [Google Scholar] [CrossRef]
- Melo-Filho, C.C.; Braga, R.C.; Muratov, E.N.; Franco, C.H.; Moraes, C.B.; Freitas-Junior, L.H.; Andrade, C.H. Discovery of New Potent Hits against Intracellular Trypanosoma cruzi by QSAR-Based Virtual Screening. Eur. J. Med. Chem. 2019, 163, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Padilla, A.; Noiva, R.; Lee, N.; Mohan, K.V.; Nakhasi, H.L.; Debrabant, A. An atypical protein disulfide isomerase from the protozoan parasite Leishmania containing a single thioredoxin-like domain. J. Biol. Chem. 2003, 278, 1872–1878. [Google Scholar] [CrossRef]
- Ware, J.M.; O’Connell, E.M.; Brown, T.; Wetzler, L.; Talaat, K.R.; Nutman, T.B.; Nash, T.E. Efficacy and Tolerability of Miltefosine in the Treatment of Cutaneous Leishmaniasis. Clin. Infect. Dis. 2021, 73, e2457–e2562. [Google Scholar] [CrossRef]
- Benaim, G.; Paniz-Mondolfi, A. Unmasking the Mechanism behind Miltefosine: Revealing the Disruption of Intracellular Ca2+ Homeostasis as a Rational Therapeutic Target in Leishmaniasis and Chagas Disease. Biomolecules 2024, 14, 406. [Google Scholar] [CrossRef]
- Facchinetti, V.; Avellar, M.M.; Nery, A.C.S.; Gomes, C.R.B.; Vasconcelos, T.R.A.; de Souza, M.V.N. An Eco-friendly, Hantzsch-Based, Solvent-Free Approach to 2-Aminothiazoles and 2-Aminoselenazoles. Synthesis 2016, 48, 437–440. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ar (p−4) | Binding Energy (∆G) | Inhibition Constant (nM) | * LogP |
|---|---|---|---|---|
| 3f | NO2 | −16.08 | 0.0019 | 4.33 |
| 3e | CN | −15.99 | 0.0024 | 4.11 |
| 3b | CH3 | −12.94 | 0.32 | 4.80 |
| 3d | Cl | −12.06 | 2.31 | 4.94 |
| 3a | H | −10.7 | 14.33 | 4.27 |
| 3c | F | −10.54 | 23.01 | 5.03 |
| Miltefosine | - | −8.21 | 951.45 | 3.44 |
| 1f | NO2 | −7.69 | 2310 | 2.35 |
| 1e | CN | −7.49 | 3580 | 2.15 |
| 1d | Cl | −6.77 | 10890 | 3.07 |
| 1b | CH3 | −6.62 | 14130 | 2.84 |
| 1a | H | −6.34 | 22590 | 2.79 |
| 2 | - | −6.22 | 27892 | 2.54 |
| 1c | F | −6.15 | 30580 | 2.46 |
| Compound | Ar (p−4) | Binding Energy (∆G) | Inhibition Constant (nM) | LogP |
|---|---|---|---|---|
| 3f | NO2 | −15.09 | 0.0021 | 4.33 |
| 3e | CN | −14.99 | 0.0024 | 4.11 |
| 3b | CH3 | −12.94 | 0.32 | 4.80 |
| 3d | Cl | −11.16 | 2.31 | 4.94 |
| 3a | H | −9.96 | 780.45 | 4.27 |
| 3c | F | −9.15 | 870.04 | 5.03 |
| Miltefosine | - | −8.21 | 951.45 | 3.44 |
 | |||
| Compound | R | Melting Point (°C) | Yield (%) |
| 1a | H | 148.7–150.0 | 90 |
| 1b | CH3 | 134.5–135.2 | 83 |
| 1c | F | 107.7–108.5 | 85 |
| 1d | Cl | 168.4–168.9 | 75 |
| 1e | CN | 177.3–177.7 | 87 |
| 1f | NO2 | 159.4–159.9 | 72 |
 | |||
| Compound | R | Melting Point (°C) | Yield (%) |
| 3a | H | 257.6–258.4 | 49 |
| 3b | CH3 | 261.2–262.1 | 45 |
| 3c | F | 270.5–271.2 | 51 |
| 3d | Cl | 262.4–263.1 | 60 |
| * 3e | CN | Complex mix | * Nq |
| 3f | NO2 | 254.4–254.8 | 54 |
| Compound | Ar (−4) | 1 LogP | 2 −∆G | 2 -KI (nM) | 3 IC50 (µM) | 3* IC90 (µM) | 4 MIC (µM) | 5 CC50 (µM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| * 3f | NO2 | 4.33 | 16.08 | 0.0019 | 22.41 | 201.69 | 0.42 | 2000.2 | 89.3 |
| * 3b | CH3 | 4.35 | 15.99 | 0.32 | 96.03 | 864.27 | 1.82 | 2016.8 | 21 |
| * 3d | Cl | 3.62 | 12.06 | 2.31 | 100.39 | 903.51 | 0.87 | 2231.6 | 22.2 |
| 1f | NO2 | 2.35 | 7.69 | 2310 | 119.50 | 1075.50 | 1.72 | 4072.6 | 18.6 |
| MTF | - | 2.68 | 8.21 | 951.45 | 132.42 | 1191.78 | 3.75 | 3556.3 | 26.9 |
| 1d | Cl | 3.07 | 6.77 | 10,890 | 347.99 | 3131.91 | 7.26 | 4425.2 | 12.7 |
| 1b | CH3 | 2.84 | 6.62 | 14,130 | 406.63 | 3659.67 | 8.04 | 4834.8 | 11.9 |
| 3a | H | 3.62 | 10.7 | 14.33 | 430.93 | 3878.37 | 124 | 2120 | 4.9 |
| 3c | F | 4.25 | 10.54 | 23.01 | 644.25 | 5798.25 | 29.66 | 1893.2 | 2.9 |
| 1a | H | 2.39 | 6.34 | 22,590 | 1446.43 | 13,017.87 | 70.93 | 5100 | 3.5 |
| 2 | H | 1.68 | 6.22 | 27,892 | 1661.45 | 14,953.05 | 203.88 | 3224.2 | 1.9 |
| 1c | F | 2.56 | 6.15 | 30,580 | 2141.54 | 19,273.86 | 64.34 | 4664 | 2.2 |
| Compound | R | LD50 (mg/kg) |
|---|---|---|
| 3a | H | >2000 |
| 3b | CH3 | >2000 |
| 3c | F | 500–1000 |
| 3d | Cl | 500 |
| 3f | NO2 | >2000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Ramírez-Sandoval, C.; Campos-Aldrete, M.E.; Meléndez-Camargo, M.E. Novel Thiazolylimidazole Hybrids as Promising Antileishmanial Agents: Rational Design and Biological Evaluation. Pathogens 2026, 15, 544. https://doi.org/10.3390/pathogens15050544
Ramírez-Sandoval C, Campos-Aldrete ME, Meléndez-Camargo ME. Novel Thiazolylimidazole Hybrids as Promising Antileishmanial Agents: Rational Design and Biological Evaluation. Pathogens. 2026; 15(5):544. https://doi.org/10.3390/pathogens15050544
Chicago/Turabian StyleRamírez-Sandoval, Cristoper, María Elena Campos-Aldrete, and María Estela Meléndez-Camargo. 2026. "Novel Thiazolylimidazole Hybrids as Promising Antileishmanial Agents: Rational Design and Biological Evaluation" Pathogens 15, no. 5: 544. https://doi.org/10.3390/pathogens15050544
APA StyleRamírez-Sandoval, C., Campos-Aldrete, M. E., & Meléndez-Camargo, M. E. (2026). Novel Thiazolylimidazole Hybrids as Promising Antileishmanial Agents: Rational Design and Biological Evaluation. Pathogens, 15(5), 544. https://doi.org/10.3390/pathogens15050544