
Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots


Abstract
1. Introduction
2. Results and Discussion
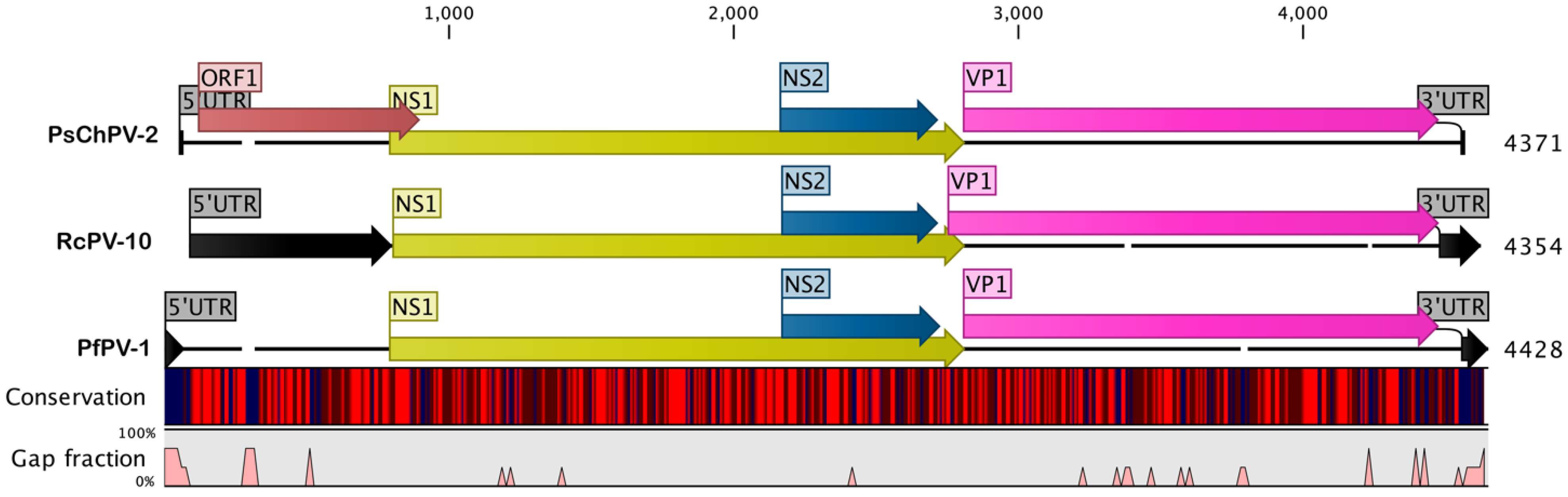
2.1. Genome of PsChPV-2
2.2. Comparative Analyses of PsChPV-2
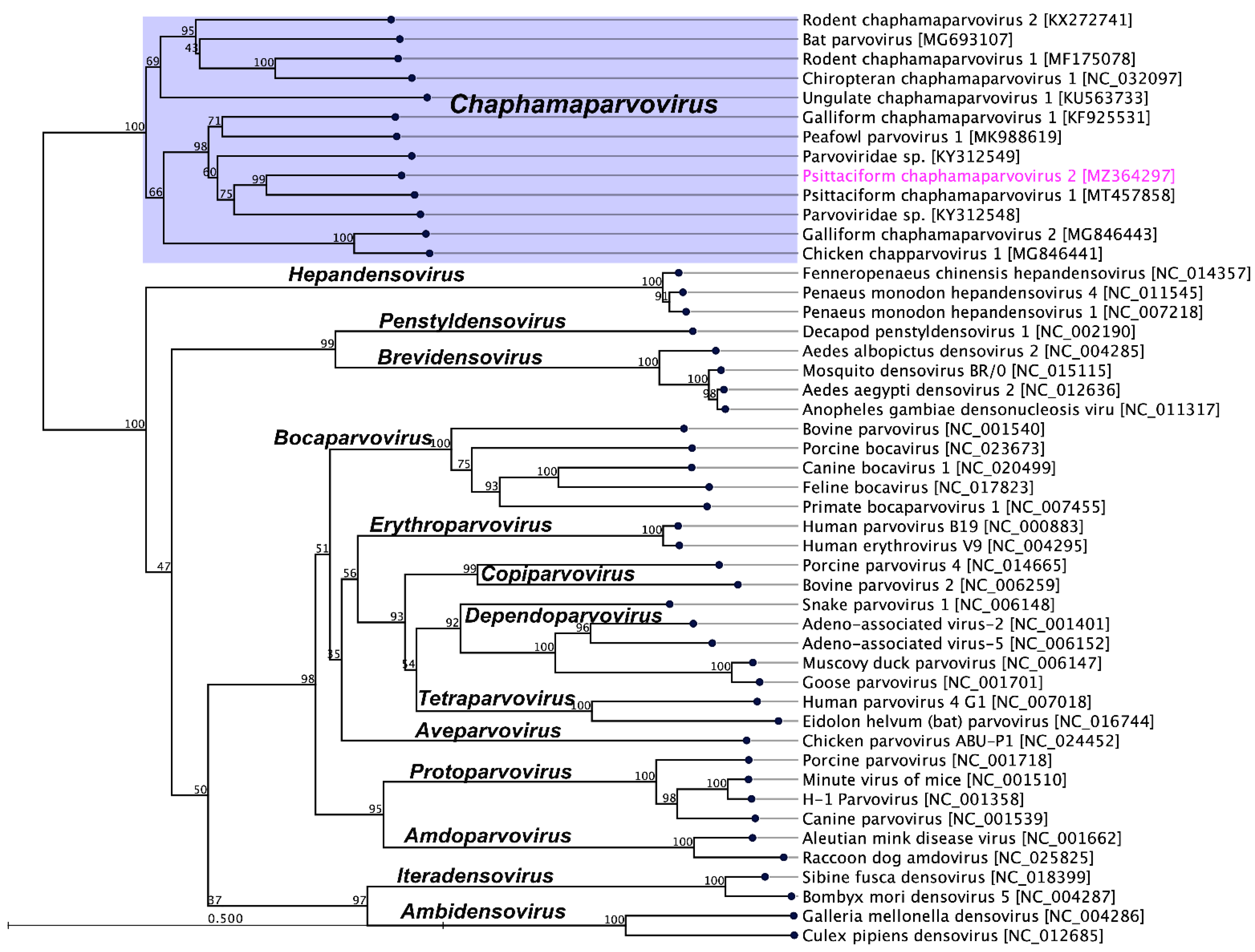
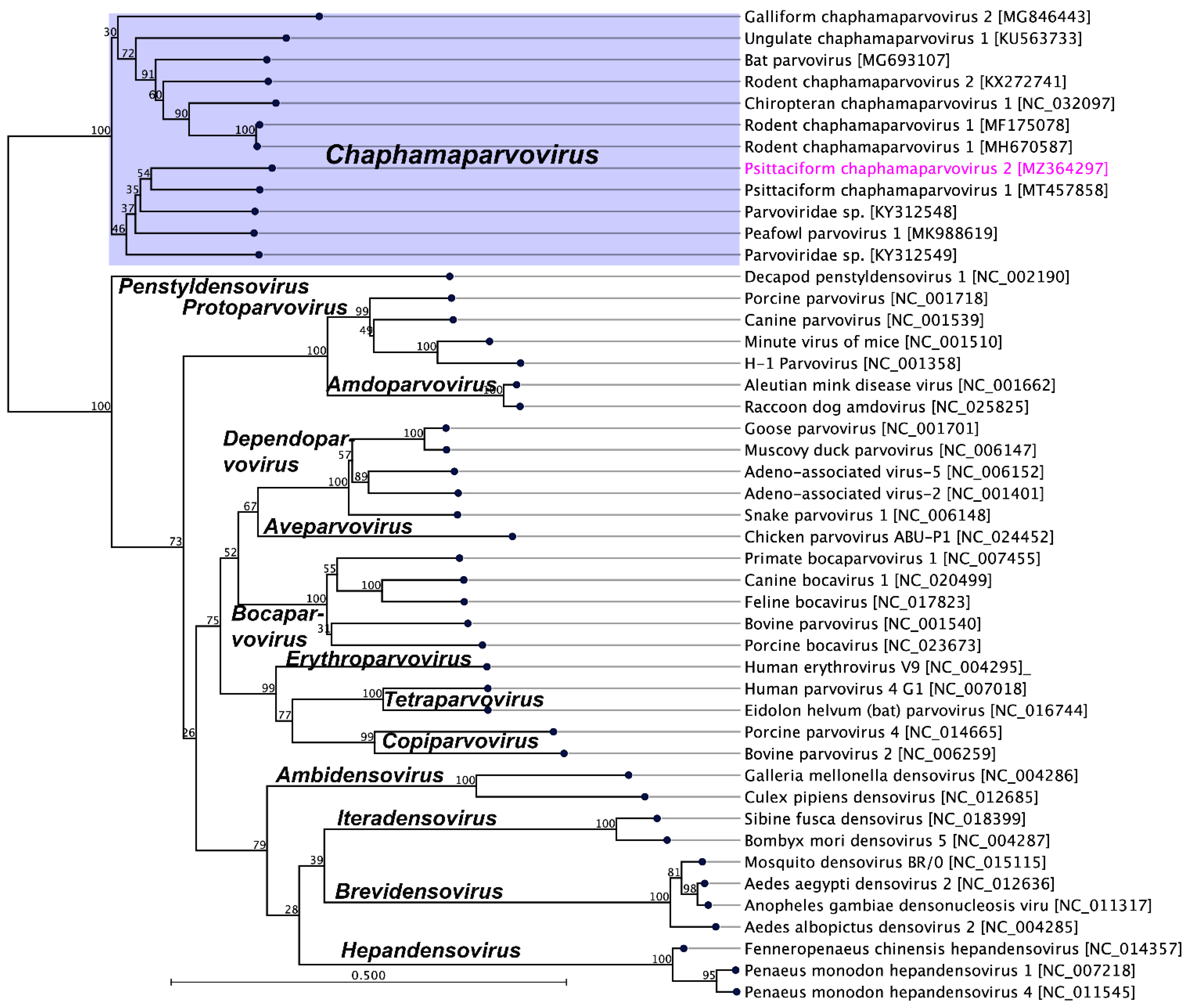
2.3. Evolutionary Relationships of PsChPV-2
3. Materials and Methods
3.1. Sampling and Ethical Approval
3.2. Virus Enrichment and Virus Nucleic Acid Extraction
3.3. Next-Generation Sequencing
3.4. Bioinformatic Analyses
3.5. Comparative Genomics and Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Gulland, F.; Simeone, C.; Deng, X.; Delwart, E. Sesavirus: Prototype of a new parvovirus genus in feces of a sea lion. Virus Genes 2015, 50, 134–136. [Google Scholar] [CrossRef][Green Version]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the family Parvoviridae: A revised taxonomy independent of the canonical approach based on host association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel circular single-stranded DNA virus from turkey faeces. Arch. Virol. 2014, 159, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-coloured fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.S.; Eden, J.S.; Hall, J.; Shi, M.; Rose, K.; Holmes, E.C. Metatranscriptomic Analysis of Virus Diversity in Urban Wild Birds with Paretic Disease. J. Virol. 2020, 94, e00606–e00620. [Google Scholar] [CrossRef]
- Souza, W.M.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef]
- Kivovich, V.; Gilbert, L.; Vuento, M.; Naides, S.J. The putative metal coordination motif in the endonuclease domain of human Parvovirus B19 NS1 is critical for NS1 induced S phase arrest and DNA damage. Int. J. Biol. Sci. 2012, 8, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Liu, X.; Li, Y.; Chen, J.; Zhang, J.; Wang, X.; Shen, S.; Wang, H.; Deng, F.; et al. Genomic and transcriptional analyses of novel parvoviruses identified from dead peafowl. Virology 2020, 539, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. mBio 2018, 9, e01354-17. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Z.; Wang, Y.; Li, W.; Fu, X.; Lin, Y.; Shen, Q.; Wang, X.; Wang, H.; Zhang, W. A novel rodent Chapparvovirus in feces of wild rats. Virol. J. 2016, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef]
- Sarker, S. Metagenomic detection and characterisation of multiple viruses in apparently healthy Australian Neophema birds. Sci. Rep. 2021, 11, 20915. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Das, S.; Lavers, J.L.; Hutton, I.; Helbig, K.; Imbery, J.; Upton, C.; Raidal, S.R. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genom. 2017, 18, 298. [Google Scholar] [CrossRef]
- Sarker, S.; Roberts, H.K.; Tidd, N.; Ault, S.; Ladmore, G.; Peters, A.; Forwood, J.K.; Helbig, K.; Raidal, S.R. Molecular and microscopic characterization of a novel Eastern Grey Kangaroopox Virus genome directly from a clinical sample. Sci. Rep. 2017, 7, 16472. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Batinovic, S.; Talukder, S.; Das, S.; Park, F.; Petrovski, S.; Forwood, J.K.; Helbig, K.J.; Raidal, S.R. Molecular characterisation of a novel pathogenic avipoxvirus from the Australian magpie (Gymnorhina tibicen). Virology 2020, 540, 1–16. [Google Scholar] [CrossRef]
- Sarker, S.; Das, S.; Helbig, K.; Peters, A.; Raidal, S.R. Genome sequence of an Australian strain of canid alphaherpesvirus 1. Aust. Vet. J. 2018, 96, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Bowden, T.R.; Boyle, D.B. Genomic characterisation of a novel avipoxvirus, magpiepox virus 2, from an Australian magpie (Gymnorhina tibicen terraereginae). Virology 2021, 562, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Athukorala, A.; Raidal, S.R. Molecular characterisation of a novel pathogenic avipoxvirus from an Australian passerine bird, mudlark (Grallina cyanoleuca). Virology 2021, 554, 66–74. [Google Scholar] [CrossRef]
- Sarker, S.; Athukorala, A.; Bowden, T.R.; Boyle, D.B. Genomic Characterisation of a Novel Avipoxvirus Isolated from an Endangered Yellow-Eyed Penguin (Megadyptes antipodes). Viruses 2021, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Chapparvoviruses | Host | GenBank Accession Numbers | Abbreviation | Genome Identity (%) | NS1 | VP1 | References | ||
|---|---|---|---|---|---|---|---|---|---|
| nt | aa | nt | aa | ||||||
| Psittaciform chaphamaparvovirus 2 | Neophema elegans/ N. splendida | MZ364297 | PsChPV-2 | This study | |||||
| Psittaciform chaphamaparvovirus 1 | Trichoglossus moluccanus | MT457858 | PsChPV-1 | 60.6 | 65.3 | 56.2 | 54.6 | 47.7 | [9] |
| Peafowl parvovirus 1 | Pavo cristatus | MK988619 | PfPV-1 | 53.8 | 59.9 | 49.2 | 49.8 | 43.4 | [12] |
| Galliform chaphamaparvovirus 1 | Meleagris gallopavo | KF925531 | GaChV-1 | 52.7 | 57.9 | 43.9 | [5] | ||
| Chicken chapparvovirus 1 | Gallus gallus | MG846441 | CChV-1 | 50.0 | 50.2 | 36.4 | unpublished * | ||
| Galliform chaphamaparvovirus 2 | Gallus gallus | MG846443 | GaChV-2 | 45.8 | 48.8 | 34.5 | 45.6 | 43.7 | unpublished * |
| Parvoviridae sp. | Grus japonensis | KY312548 | RcPV-9 | 55.6 | 59.4 | 49.7 | 53.1 | 40.5 | unpublished * |
| Parvoviridae sp. | Grus japonensis | KY312549 | RcPV-10 | 53.5 | 59.5 | 46.8 | 52.8 | 39.1 | unpublished * |
| Rodent chaphamaparvovirus 1 | Mus musculus | MF175078 | RoChV-1 | 45.4 | 47.6 | 36.7 | 47.0 | 39.7 | [13] |
| Chiropteran chaphamaparvovirus 1 | Desmodus rotundus | NC_032097 | ChiCPV-1 | 46.0 | 49.2 | 34.0 | 47.0 | 39.6 | [10] |
| Bat parvovirus | Eidolon helvum | MG693107 | BtPV | 46.4 | 48.5 | 33.4 | 46.0 | 39.7 | [14] |
| Rodent chaphamaparvovirus 2 | wild rat | KX272741 | RoChV-2 | 44.2 | 47.8 | 35.8 | 43.5 | 37.7 | [15] |
| Ungulate chaphamaparvovirus 1 | swine | KU563733 | UChPPV-1 | 41.1 | 43.4 | 31.7 | 42.0 | 35.2 | [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarker, S. Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathogens 2021, 10, 1559. https://doi.org/10.3390/pathogens10121559
Sarker S. Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathogens. 2021; 10(12):1559. https://doi.org/10.3390/pathogens10121559
Chicago/Turabian StyleSarker, Subir. 2021. "Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots" Pathogens 10, no. 12: 1559. https://doi.org/10.3390/pathogens10121559
APA StyleSarker, S. (2021). Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathogens, 10(12), 1559. https://doi.org/10.3390/pathogens10121559