
Highly Specific Sigma Receptor Ligands Exhibit Anti-Viral Properties in SARS-CoV-2 Infected Cells

 , , , ,
, , , ,  ,
,  , , and
, , and
Abstract
:1. Introduction
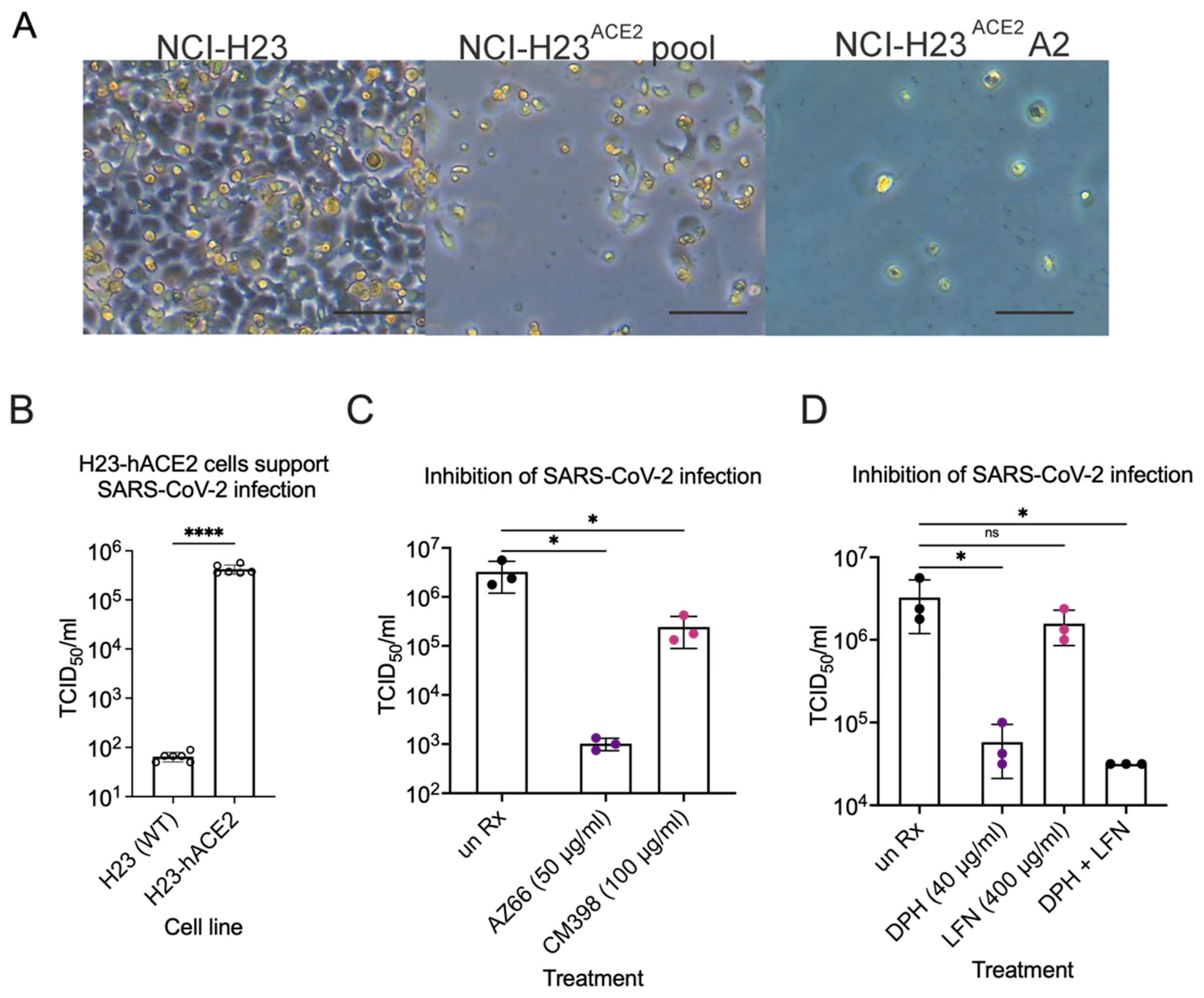
2. Results
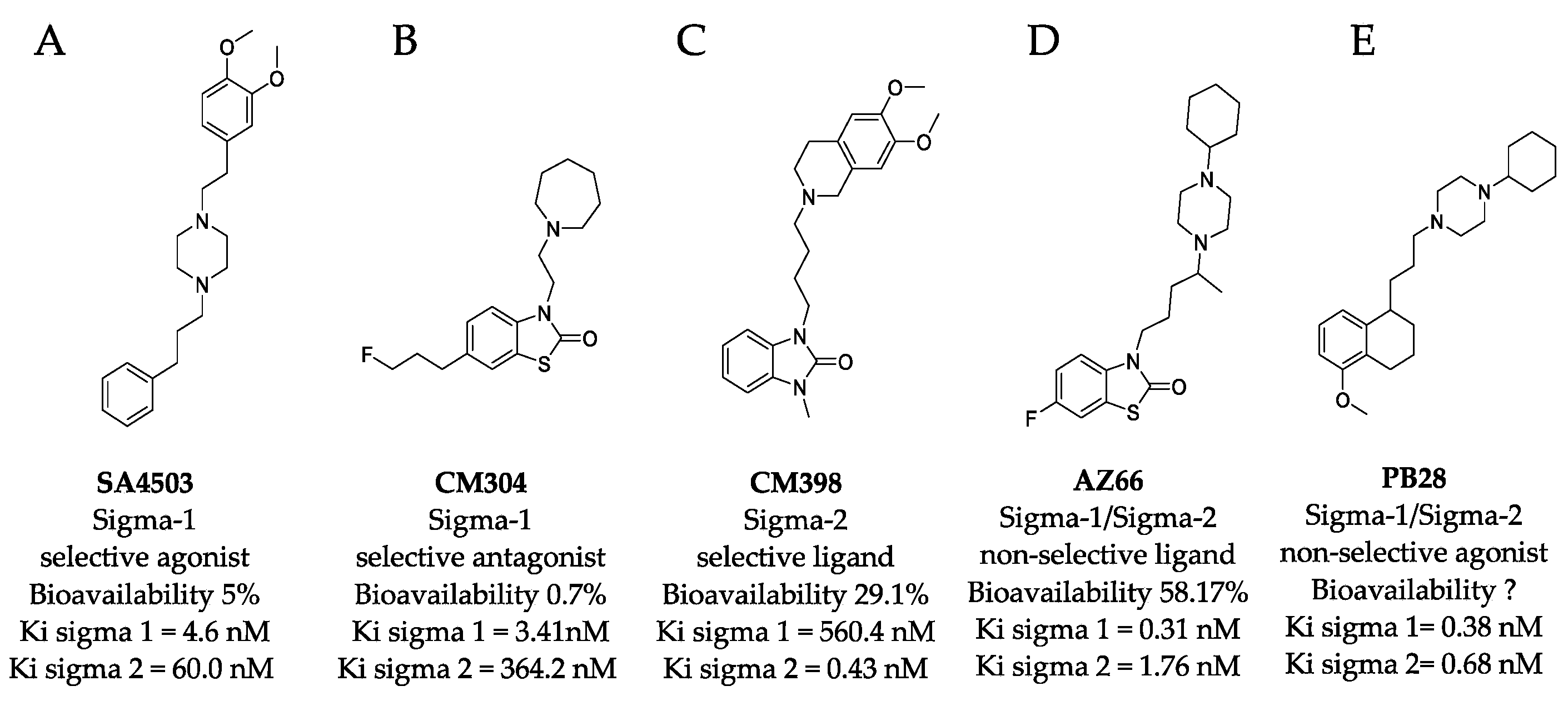
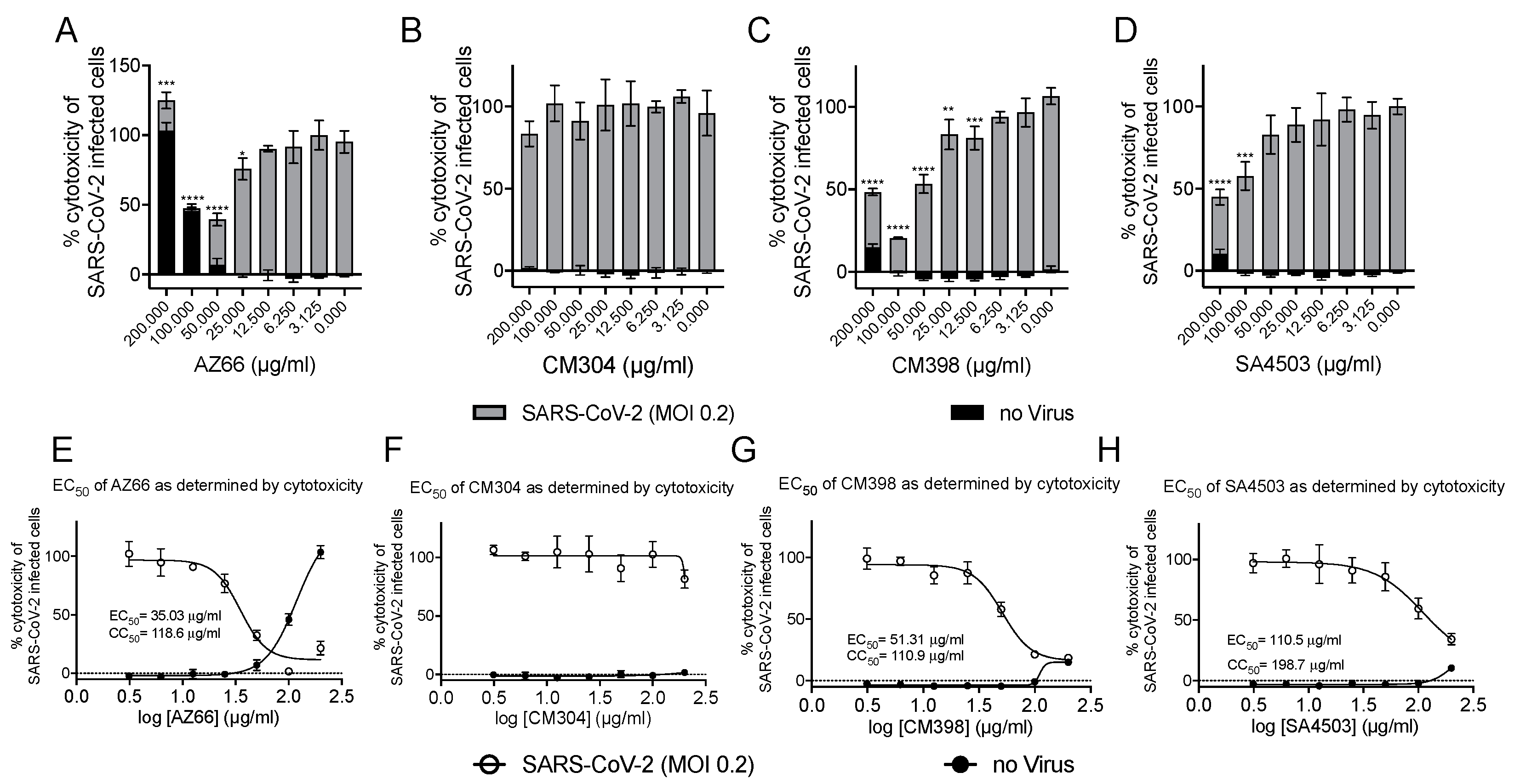
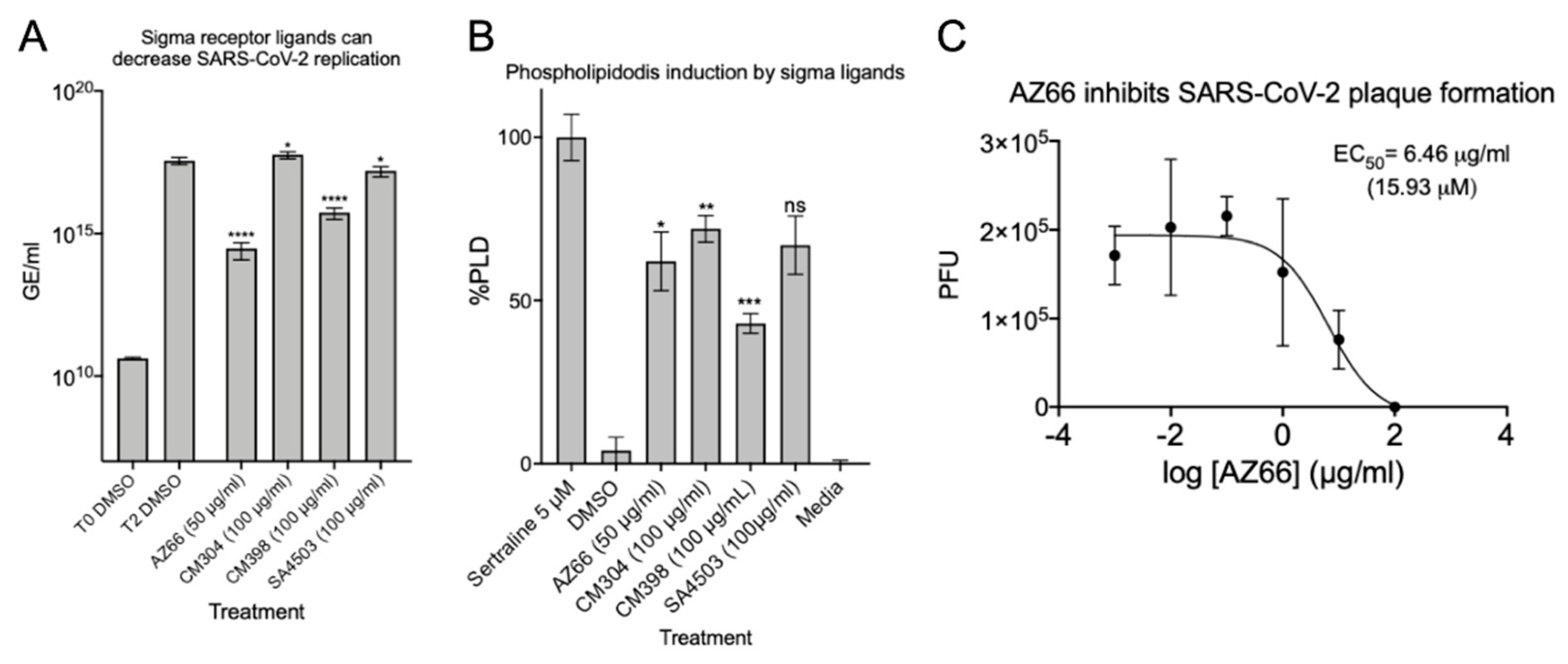
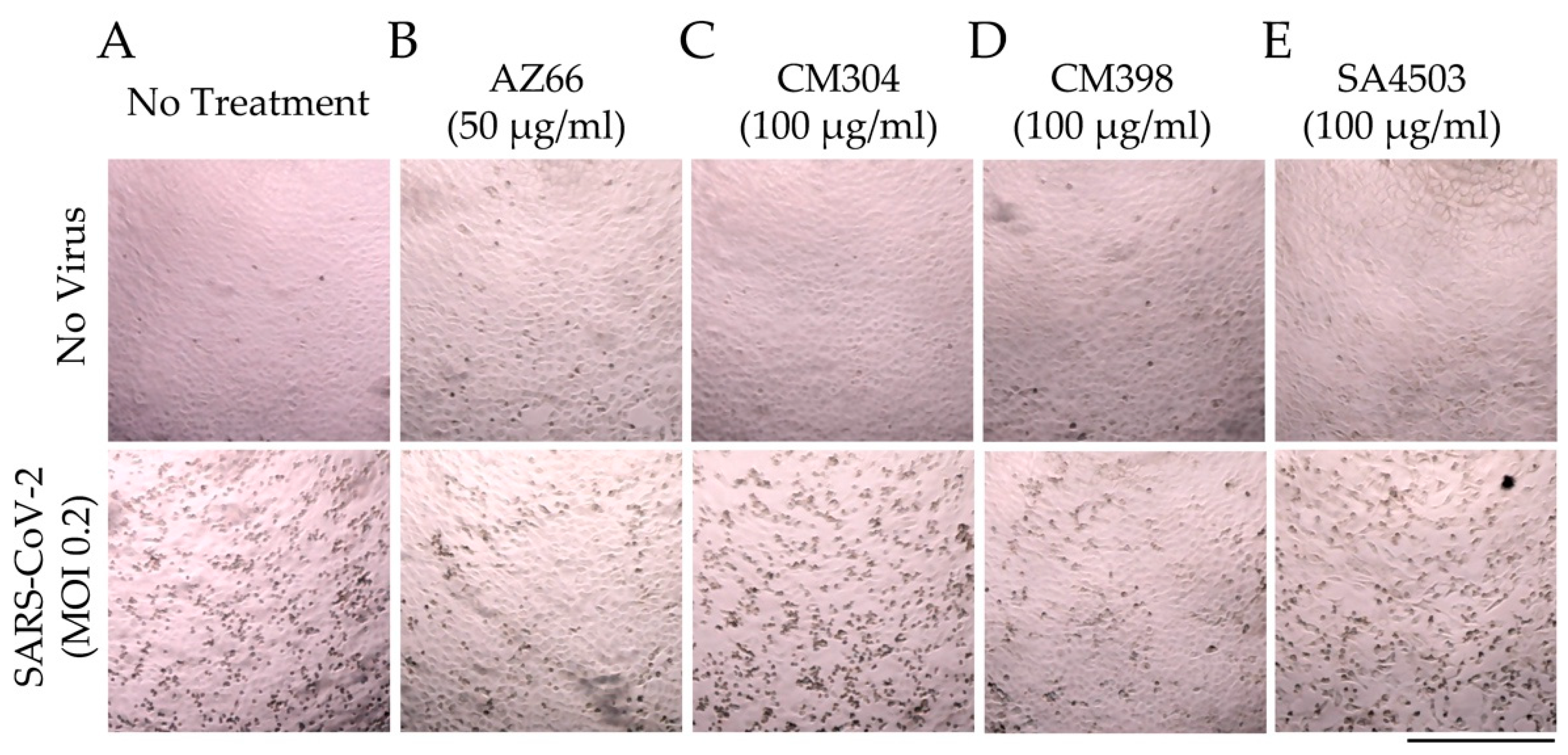
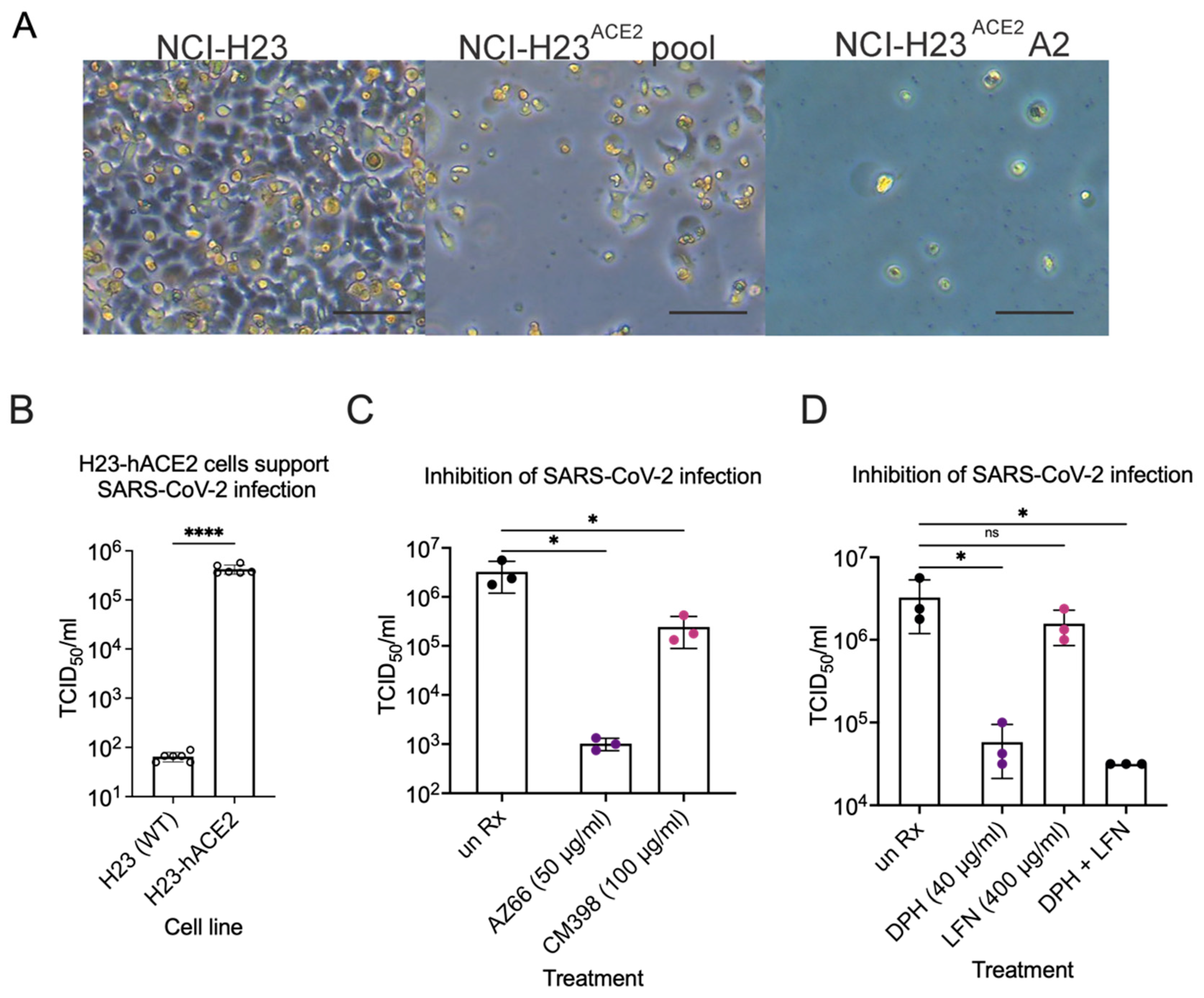
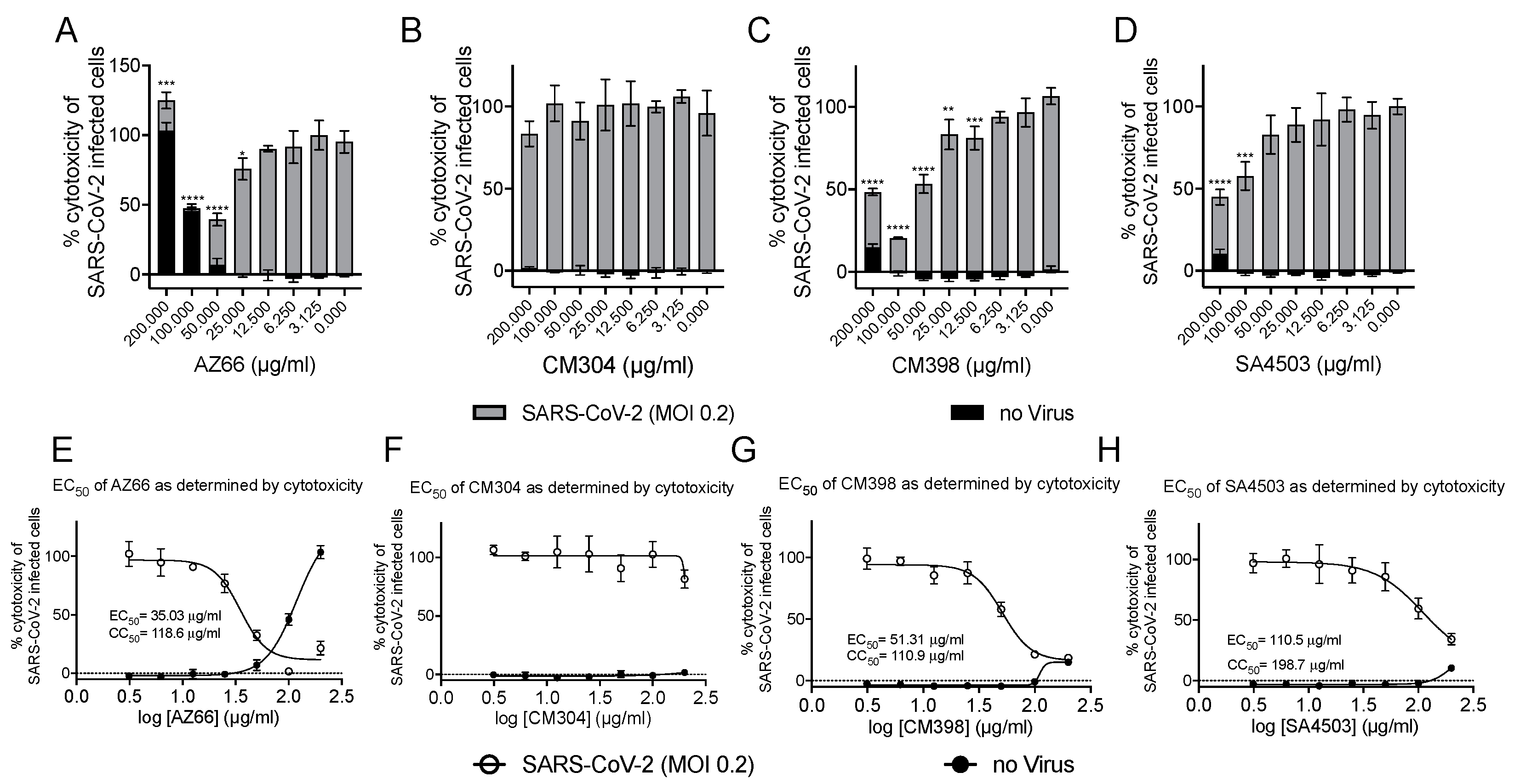
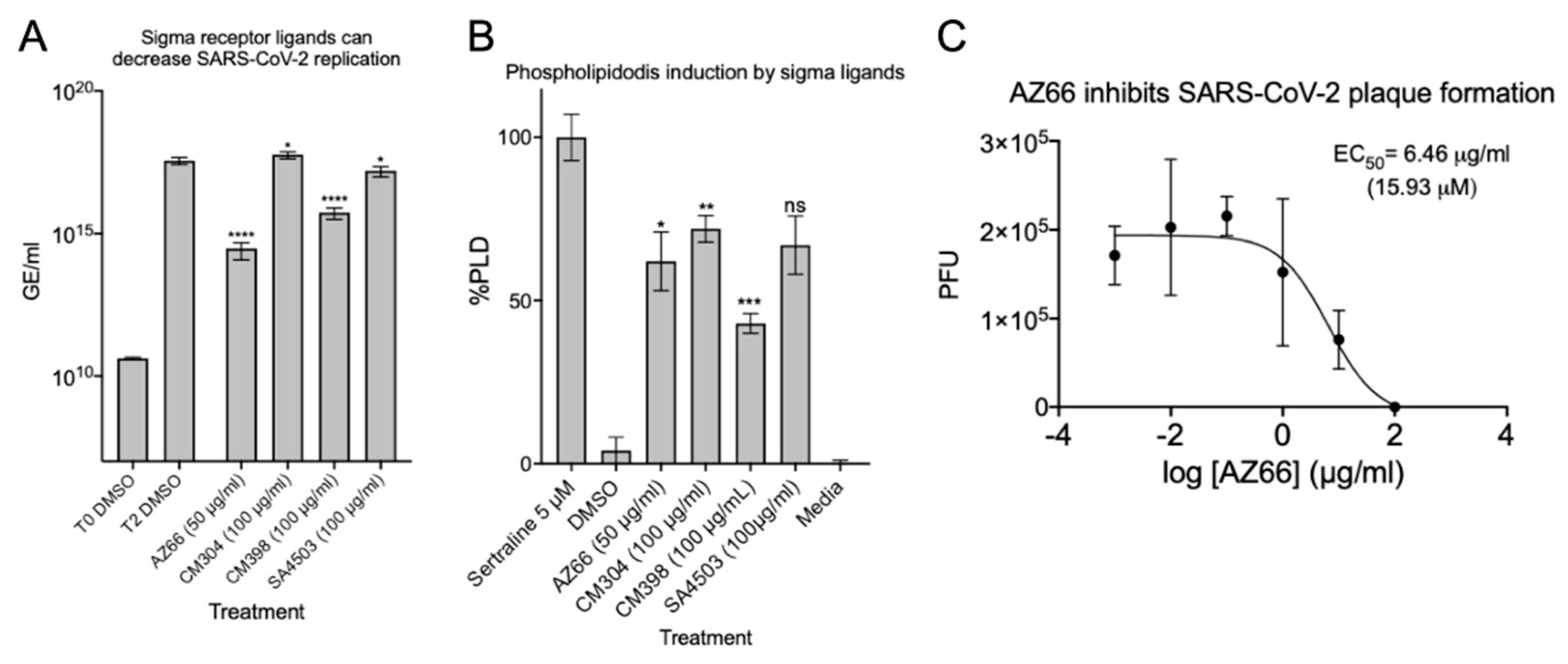
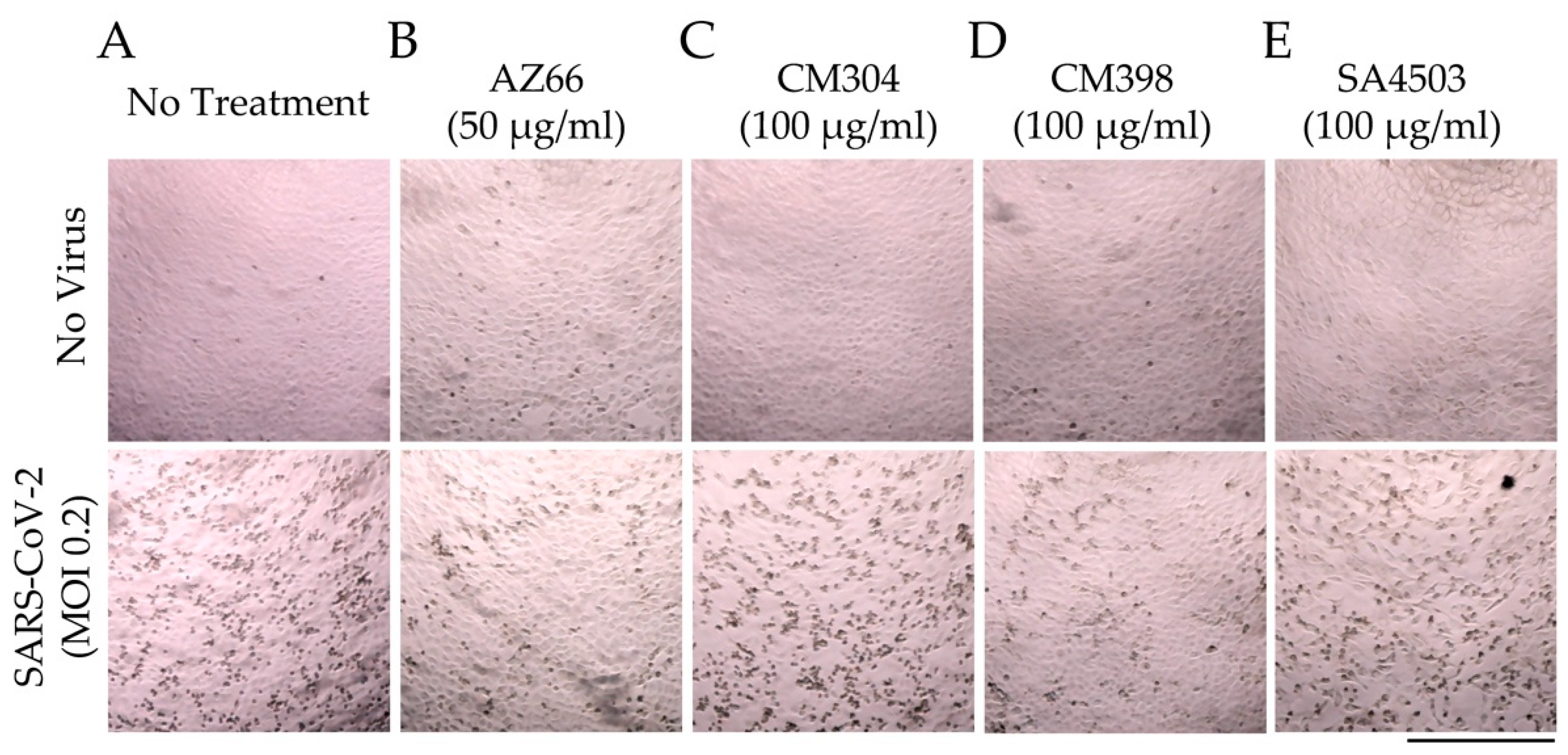
2.1. Sigma Ligands Inhibit SARS-CoV-2-Mediated Cell Death, Intracellular Replication, and Infectivity
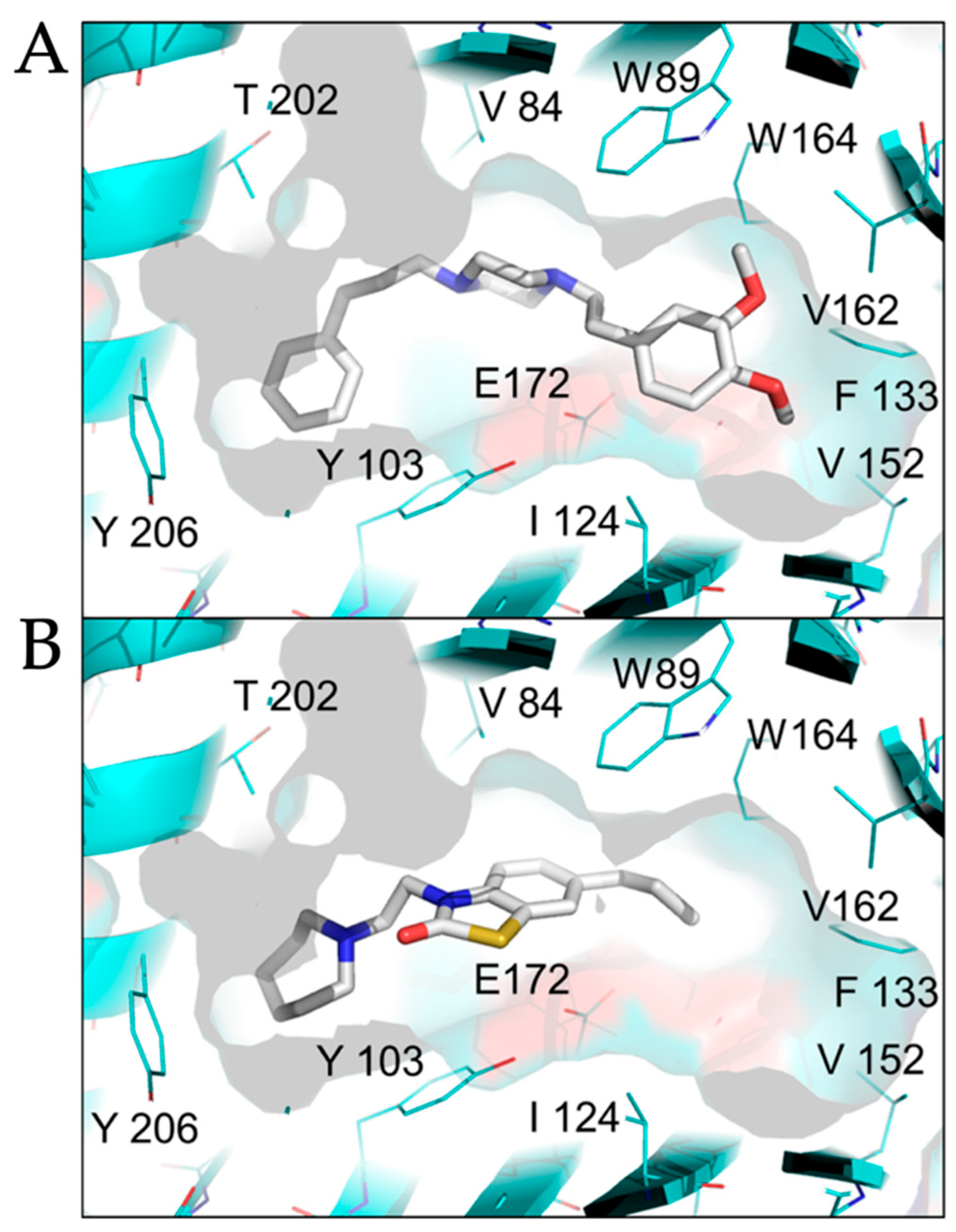
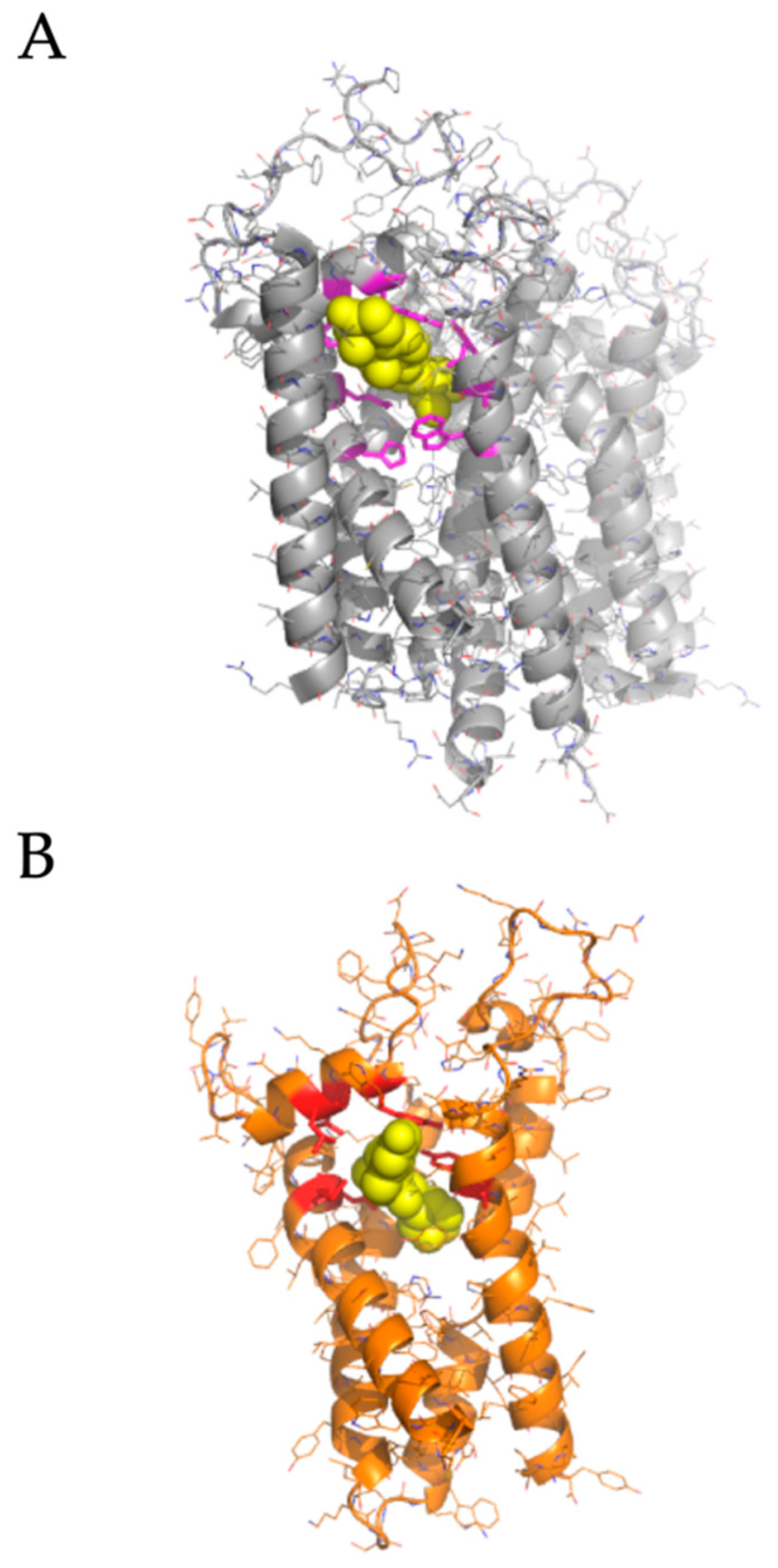
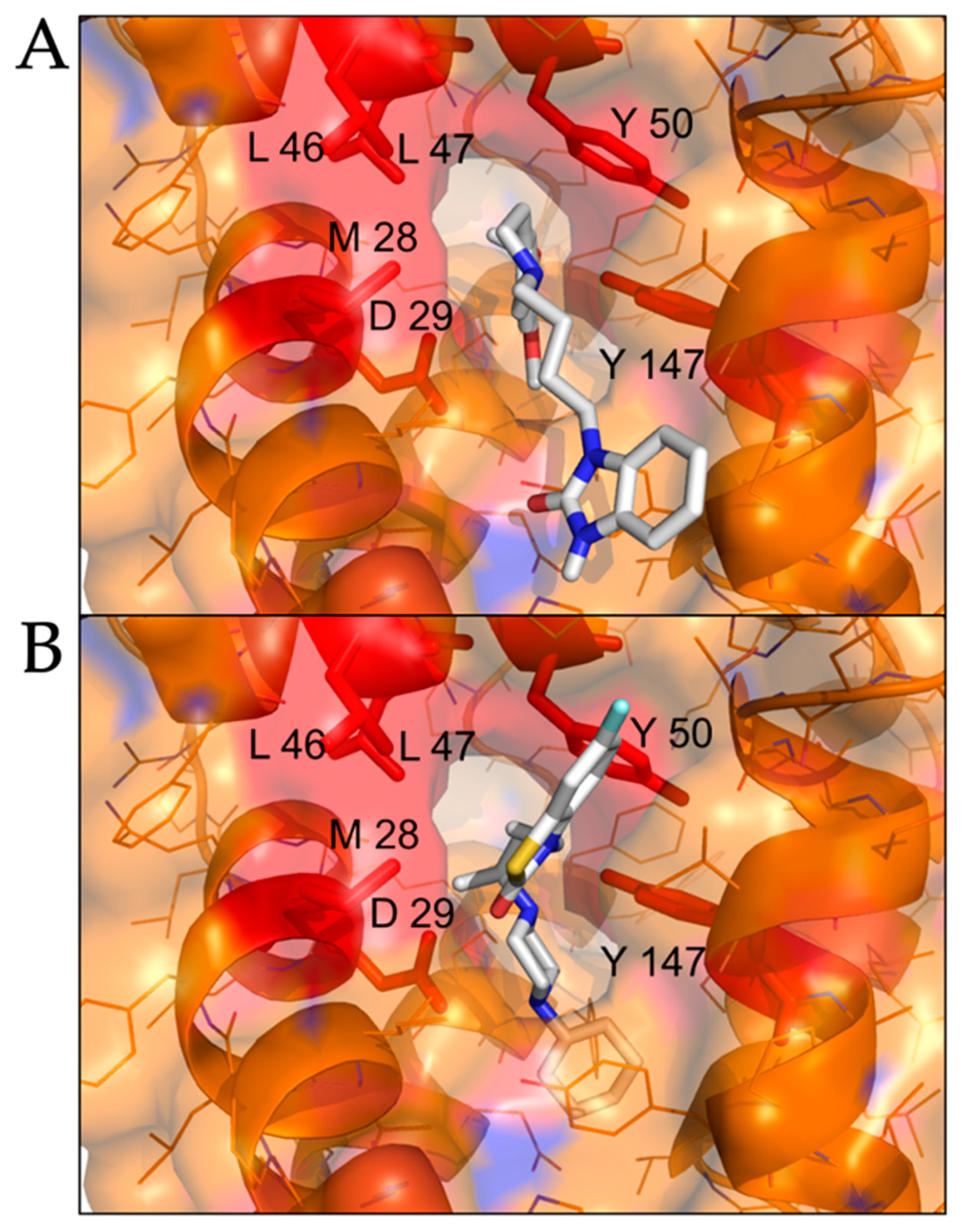
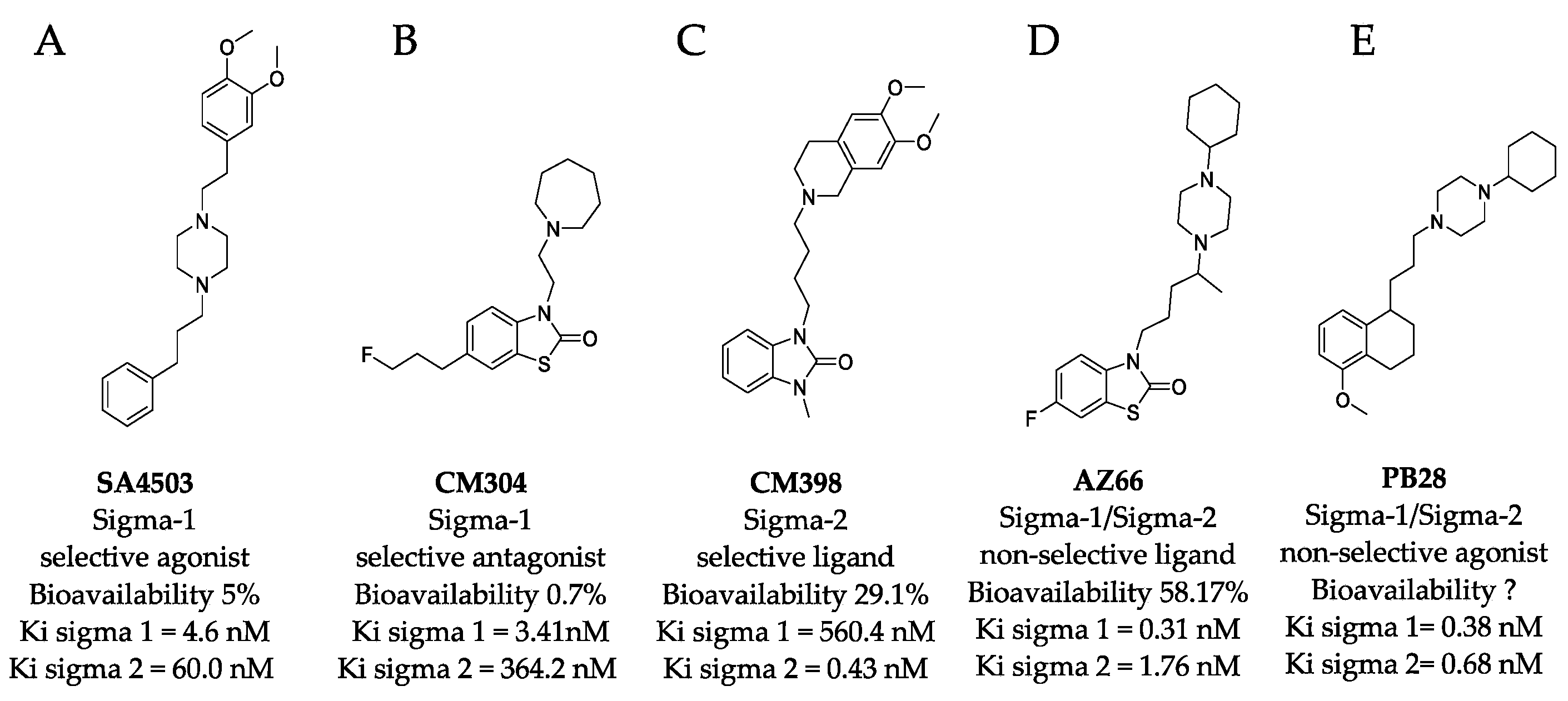
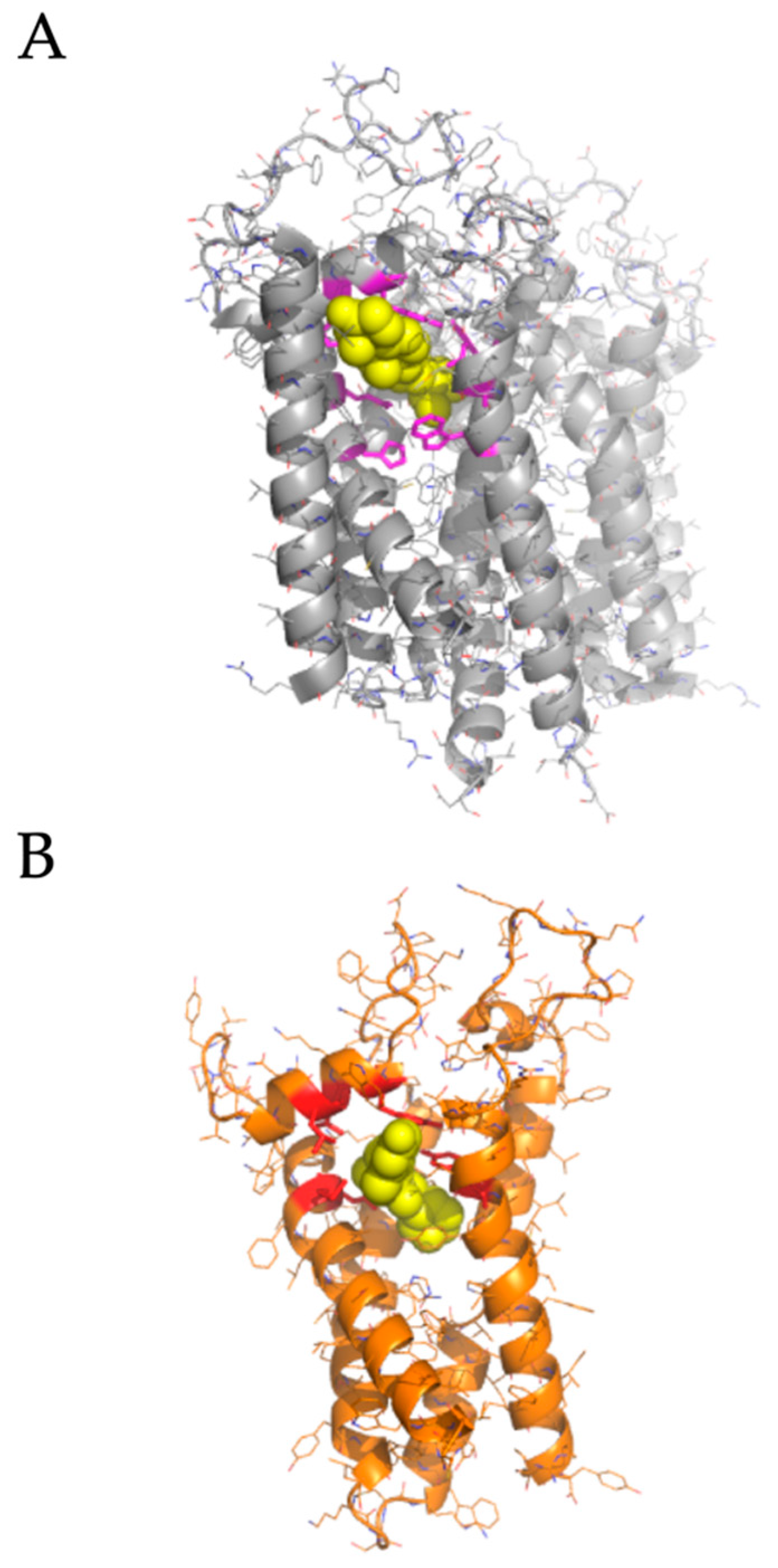
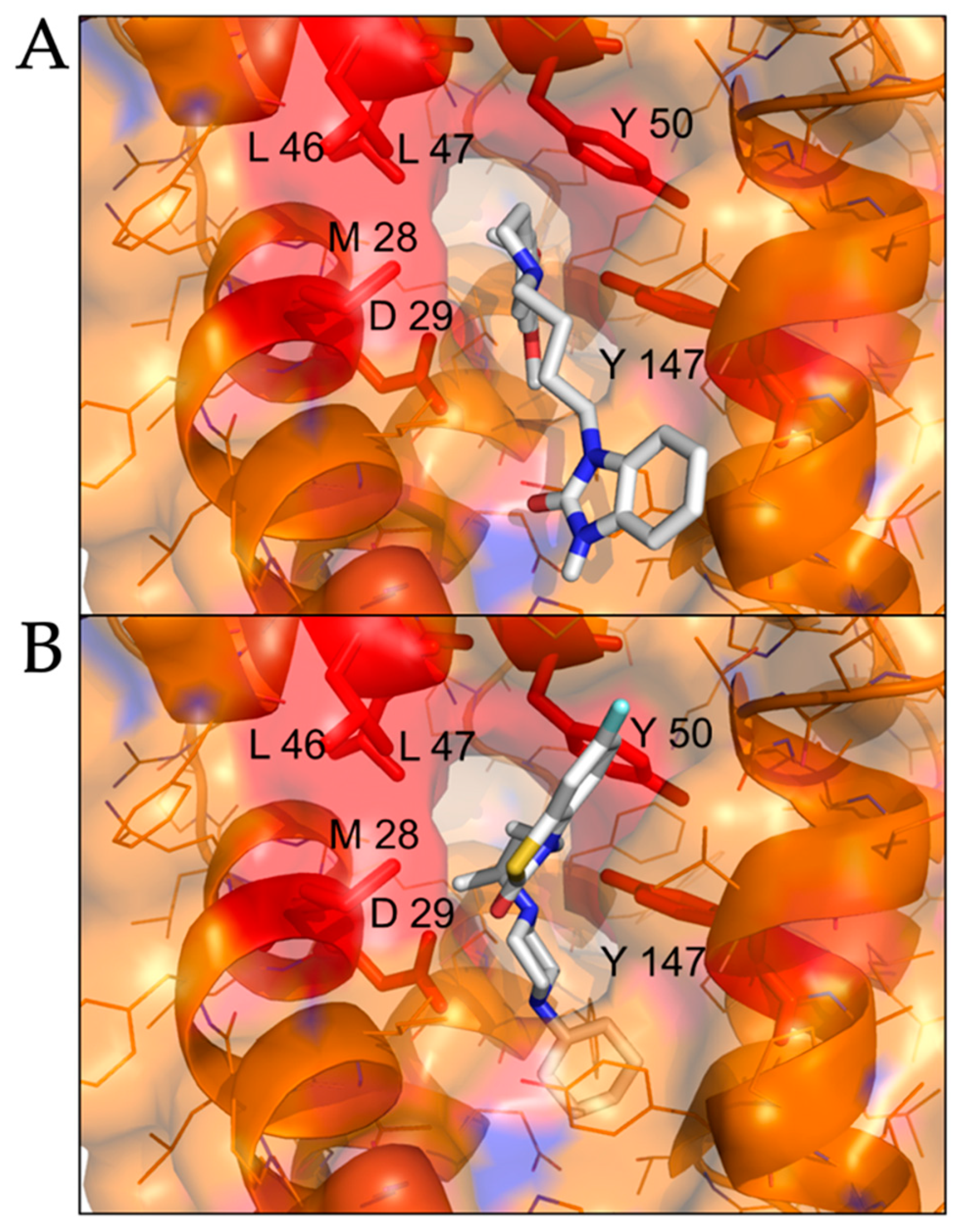
2.2. Modeled Structural Interactions between Sigma Receptors and Ligands Provides a Basis for Antiviral Drug Optimization
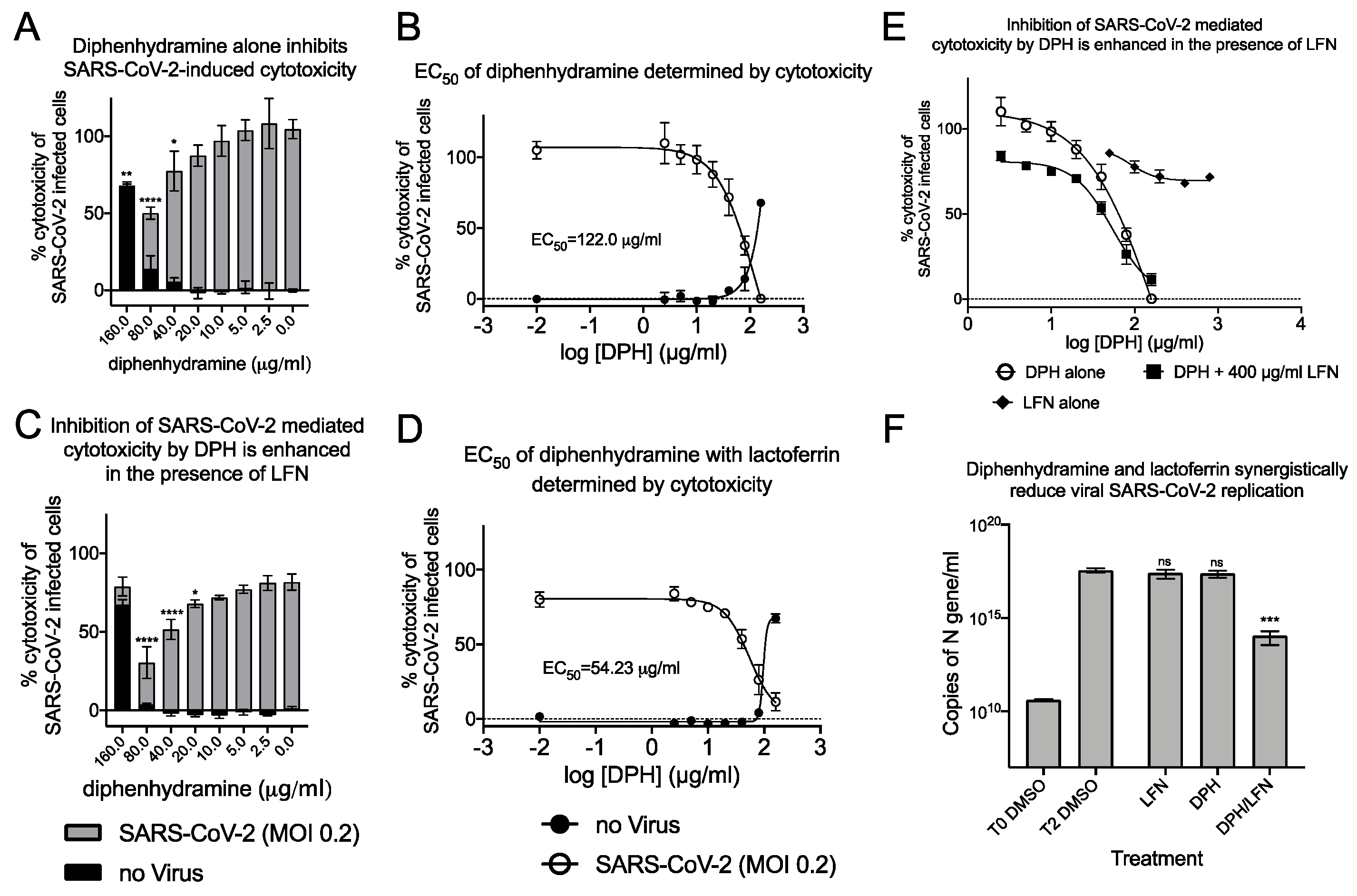
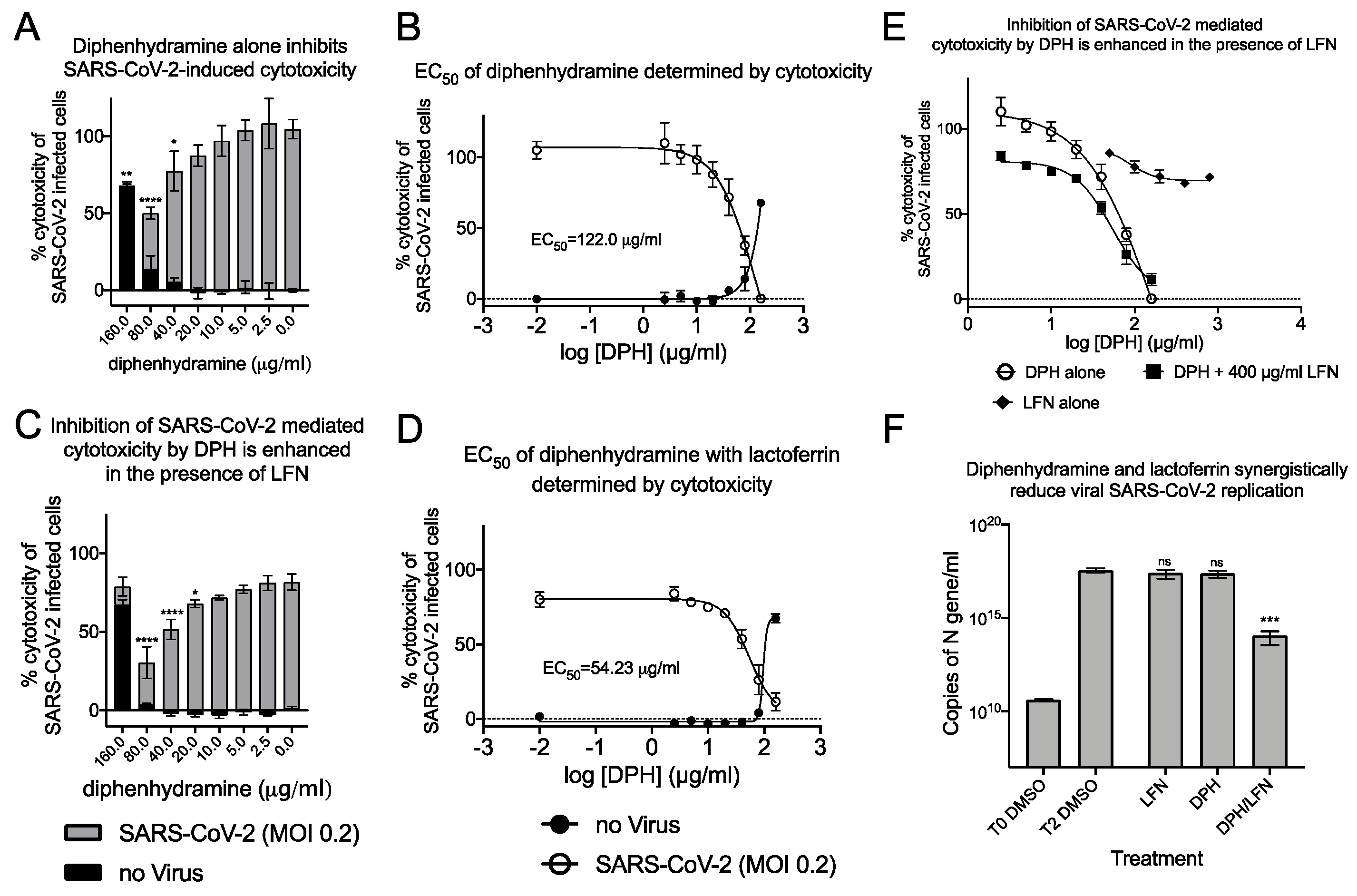
2.3. Synergistic Antiviral Activity by Combining a Sigma Receptor Ligand with Lactoferrin
2.4. Sigma Ligands Inhibit Infectious Particle Production in Human Lung Cells
3. Discussion and Conclusions
4. Materials and Methods
4.1. Sigma Ligands and Other Drugs Used in this Study
4.2. Virus Culture Methods
4.3. Quantitation of Virus Replication by qPCR
4.4. Sigma Ligand Cytotoxicity Reduction Assays
4.5. Plaque Reduction Assay
4.6. Generation of ACE-2 Lentivirus Particles
4.7. ACE2 Transduction of NCI-H23 Cells and Monoclonal Cell Selection
4.8. Analysis of Cell Surface ACE2 by Flow Cytometry
4.9. TCID50 Assays in H23 Cells
4.10. Inhibitory Concentration and Effective Concentration Calculations
4.11. Molecular Docking of Sigma Receptor Ligands
4.12. Phospholipidosis Assay
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Joshi, M.; Degani, M.S. Tackling SARS-CoV-2: Proposed targets and repurposed drugs. Futur. Med. Chem. 2020, 12, 1579–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Poduri, R.; Joshi, G.; Jagadeesh, G. Drugs targeting various stages of the SARS-CoV-2 life cycle: Exploring promising drugs for the treatment of covid-19. Cell Signal. 2020, 74, 109721. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Reznikov, L.R.; Norris, M.H.; Vashisht, R.; Bluhm, A.P.; Li, D.; Liao, Y.-S.J.; Brown, A.; Butte, A.J.; Ostrov, D.A. Identification of antiviral antihistamines for COVID-19 repurposing. Biochem. Biophys. Res. Commun. 2021, 538, 173–179. [Google Scholar] [CrossRef]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef]
- Vela, J.M. Repurposing Sigma-1 Receptor Ligands for COVID-19 Therapy? Front. Pharmacol. 2020, 11, 582310. [Google Scholar] [CrossRef]
- Abate, C.; Niso, M.; Berardi, F. Sigma-2 receptor: Past, present and perspectives on multiple therapeutic exploitations. Futur. Med. Chem. 2018, 10, 1997–2018. [Google Scholar] [CrossRef]
- Zeng, C.; Riad, A.; Mach, R.H. The Biological Function of Sigma-2 Receptor/TMEM97 and Its Utility in PET Imaging Studies in Cancer. Cancers 2020, 12, 1877. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zeng, C.; Wang, C.; Sun, M.; Yin, D.; Sun, T. Sigma-2 Receptor—A Potential Target for Cancer/Alzheimer’s Disease Treatment via Its Regulation of Cholesterol Homeostasis. Molecules 2020, 25, 5439. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, S.-Z.; Yao, Y.-H.; Xiang, Y.; Ma, X.-Y.; Wei, X.-L.; Yan, H.-T.; Liu, X.-Y. Sigma-1 receptor agonist increases axon outgrowth of hippocampal neurons via voltage-gated calcium ions channels. CNS Neurosci. Ther. 2017, 23, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, K.; Ishiwata, K.; Tajima, H.; Ishii, S.-I.; Matsuno, K.; Homma, Y.; Senda, M. In vivo evaluation of [11C]SA4503 as a PET ligand for mapping CNS sigma1 receptors. Nucl. Med. Biol. 2000, 27, 255–261. [Google Scholar] [CrossRef]
- Matsuno, K.; Nakazawa, M.; Okamoto, K.; Kawashima, Y.; Mita, S. Binding properties of SA4503, a novel and selective sigma 1 receptor agonist. Eur J. Pharmacol. 1996, 306, 271–279. [Google Scholar] [CrossRef]
- Cirino, T.J.; Eans, S.O.; Medina, J.M.; Wilson, L.L.; Mottinelli, M.; Intagliata, S.; McCurdy, C.R.; McLaughlin, J.P. Characterization of Sigma 1 Receptor Antagonist CM-304 and Its Analog, AZ-66: Novel Therapeutics Against Allodynia and Induced Pain. Front. Pharmacol. 2019, 10, 678. [Google Scholar] [CrossRef] [Green Version]
- Intagliata, S.; Sharma, A.; King, T.; Mesangeau, C.; Seminerio, M.; Chin, F.T.; Wilson, L.L.; Matsumoto, R.R.; McLaughlin, J.P.; Avery, B.A.; et al. Discovery of a Highly Selective Sigma-2 Receptor Ligand, 1-(4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-3-methyl-1H-benzo[d]imidazol-2(3H)-one (CM398), with Drug-Like Properties and Antinociceptive Effects In Vivo. AAPS J. 2020, 22, 94. [Google Scholar] [CrossRef]
- Jamalapuram, S.; Vuppala, P.K.; Abdelazeem, A.H.; McCurdy, C.R.; Avery, B.A. Ultra-performance liquid chromatography tandem mass spectrometry method for the determination of AZ66, a sigma receptor ligand, in rat plasma and its application to in vivo pharmacokinetics. Biomed. Chromatogr. 2013, 27, 1034–1040. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-T.; Kaushal, N.; Shaikh, J.; Wilson, L.L.; Mésangeau, C.; McCurdy, C.R.; Matsumoto, R.R. A Novel Substituted Piperazine, CM156, Attenuates the Stimulant and Toxic Effects of Cocaine in Mice. J. Pharmacol. Exp. Ther. 2010, 333, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Mésangeau, C.; Narayanan, S.; Green, A.M.; Shaikh, J.; Kaushal, N.; Viard, E.; Xu, Y.-T.; Fishback, J.A.; Poupaert, J.H.; Matsumoto, R.R.; et al. Conversion of a Highly Selective Sigma-1 Receptor–Ligand to Sigma-2 Receptor Preferring Ligands with Anticocaine Activity. J. Med. Chem. 2008, 51, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Seminerio, M.J.; Robson, M.J.; Abdelazeem, A.H.; Mesangeau, C.; Jamalapuram, S.; Avery, B.A.; McCurdy, C.R.; Matsumoto, R.R. Synthesis and Pharmacological Characterization of a Novel Sigma Receptor Ligand with Improved Metabolic Stability and Antagonistic Effects Against Methamphetamine. AAPS J. 2012, 14, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Shuai, L.; Wen, Z.; Wang, C.; Yan, Y.; Jiao, Z.; Guo, F.; Fu, Z.F.; Chen, H.; Bu, Z.; et al. The preclinical inhibitor GS441524 in combination with GC376 efficaciously inhibited the proliferation of SARS-CoV-2 in the mouse respiratory tract. Emerg. Microbes Infect. 2021, 10, 481–492. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, T.; Hassan, A.; Thompson, B.M.; McDonald, J.G.; Wang, J.; Li, X. Structural basis for human sterol isomerase in cholesterol biosynthesis and multidrug recognition. Nat. Commun. 2019, 10, 2452. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.; Gabrielli, M.; Costa, J.; Uttaro, A.D. Phagocytic and pinocytic uptake of cholesterol in Tetrahymena thermophila impact differently on gene regulation for sterol homeostasis. Sci. Rep. 2021, 11, 9067. [Google Scholar] [CrossRef]
- Basile, A.S.; Paul, I.A.; Mirchevich, A.; Kuijpers, G.; De Costa, B. Modulation of (+)-[3H]pentazocine binding to guinea pig cerebellum by divalent cations. Mol. Pharmacol. 1992, 42, 882–889. [Google Scholar]
- Mirabelli, C.; Wotring, J.W.; Zhang, C.J.; McCarty, S.M.; Fursmidt, R.; Pretto, C.D.; Sexton, J.Z. Morphological cell profiling of SARS-CoV-2 infection identifies drug repurposing candidates for COVID-19. bioRxiv 2020, 338, e2105815118. [Google Scholar] [CrossRef]
- Hu, Y.; Meng, X.; Zhang, F.; Xiang, Y.; Wang, J. The in vitro antiviral activity of lactoferrin against common human coronaviruses and SARS-CoV-2 is mediated by targeting the heparan sulfate co-receptor. Emerg. Microbes Infect. 2021, 10, 317–330. [Google Scholar] [CrossRef]
- Li, F.; Han, M.; Dai, P.; Xu, W.; He, J.; Tao, X.; Wu, Y.; Tong, X.; Xia, X.; Guo, W.; et al. Distinct mechanisms for TMPRSS2 expression explain organ-specific inhibition of SARS-CoV-2 infection by enzalutamide. Nat. Commun. 2021, 12, 866. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus–host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef] [Green Version]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; O’Meara, M.J.; Monel, B.; Vallet, T.; White, K.M.; Zhang, Z.; Alon, A.; et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science 2021, 373, 541–547. [Google Scholar] [CrossRef]
- Zheng, Y.; Ma, S.; Xiong, Y.; Fan, X. Efficacy and safety of direct acting antiviral regimens for hepatitis C virus and human immunodeficiency virus co-infection: Systematic review and network meta-analysis. J. Gastroenterol. Hepatol. 2020, 35, 1477–1487. [Google Scholar] [CrossRef]
- Kulemina, L.V.; Ostrov, D.A. Prediction of Off-Target Effects on Angiotensin-Converting Enzyme 2. J. Biomol. Screen. 2011, 16, 878–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, T.R.; Ekins, S. Defending Antiviral Cationic Amphiphilic Drugs That May Cause Drug-Induced Phospholipidosis. J. Chem. Inf. Model. 2021, 61, 4125–4130. [Google Scholar] [CrossRef]
- James, M.L.; Shen, B.; Zavaleta, C.; Nielsen, C.H.; Mesangeau, C.; Vuppala, P.K.; Chan, C.; Avery, B.A.; Fishback, J.A.; Matsumoto, R.R.; et al. New Positron Emission Tomography (PET) Radioligand for Imaging σ-1 Receptors in Living Subjects. J. Med. Chem. 2012, 55, 8272–8282. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.E.; Li, S.; Vizeacoumar, F.S.; Bhanumathy, K.K.; Lee, J.S.; Parameswaran, S.; Furber, L.; Abuhussein, O.; Paul, J.M.; McDonald, M.; et al. Therapeutic relevance of the protein phosphatase 2A in cancer. Oncotarget 2016, 7, 61544–61561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytotoxicity | Plaque Reduction | ||||||
|---|---|---|---|---|---|---|---|
| CC50 | EC50 | EC50 | |||||
| µg/mL | R2 | µg/mL | R2 | µg/mL | R2 | ||
| AZ66 | 127.6 (109.8–173.7) | 0.9957 | 45.14 (36.91–55.21) | 0.8874 | 6.47 (1.27–32.86) | 0.7369 | |
| CM398 | 110.9 (ND) | 0.9618 | 51.31 (43.78–61.13) | 0.9629 | |||
| SA4503 | 198.7 (ND) | 0.9401 | 110.5 (39.2–311.3) | 0.8789 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostrov, D.A.; Bluhm, A.P.; Li, D.; Khan, J.Q.; Rohamare, M.; Rajamanickam, K.; K. Bhanumathy, K.; Lew, J.; Falzarano, D.; Vizeacoumar, F.J.; et al. Highly Specific Sigma Receptor Ligands Exhibit Anti-Viral Properties in SARS-CoV-2 Infected Cells. Pathogens 2021, 10, 1514. https://doi.org/10.3390/pathogens10111514
Ostrov DA, Bluhm AP, Li D, Khan JQ, Rohamare M, Rajamanickam K, K. Bhanumathy K, Lew J, Falzarano D, Vizeacoumar FJ, et al. Highly Specific Sigma Receptor Ligands Exhibit Anti-Viral Properties in SARS-CoV-2 Infected Cells. Pathogens. 2021; 10(11):1514. https://doi.org/10.3390/pathogens10111514
Chicago/Turabian StyleOstrov, David A., Andrew P. Bluhm, Danmeng Li, Juveriya Qamar Khan, Megha Rohamare, Karthic Rajamanickam, Kalpana K. Bhanumathy, Jocelyne Lew, Darryl Falzarano, Franco J. Vizeacoumar, and et al. 2021. "Highly Specific Sigma Receptor Ligands Exhibit Anti-Viral Properties in SARS-CoV-2 Infected Cells" Pathogens 10, no. 11: 1514. https://doi.org/10.3390/pathogens10111514
APA StyleOstrov, D. A., Bluhm, A. P., Li, D., Khan, J. Q., Rohamare, M., Rajamanickam, K., K. Bhanumathy, K., Lew, J., Falzarano, D., Vizeacoumar, F. J., Wilson, J. A., Mottinelli, M., Kanumuri, S. R. R., Sharma, A., McCurdy, C. R., & Norris, M. H. (2021). Highly Specific Sigma Receptor Ligands Exhibit Anti-Viral Properties in SARS-CoV-2 Infected Cells. Pathogens, 10(11), 1514. https://doi.org/10.3390/pathogens10111514