
Molecular and Epigenetic Mechanisms Underlying Cognitive and Adaptive Responses to Stress

 , , and
, , and 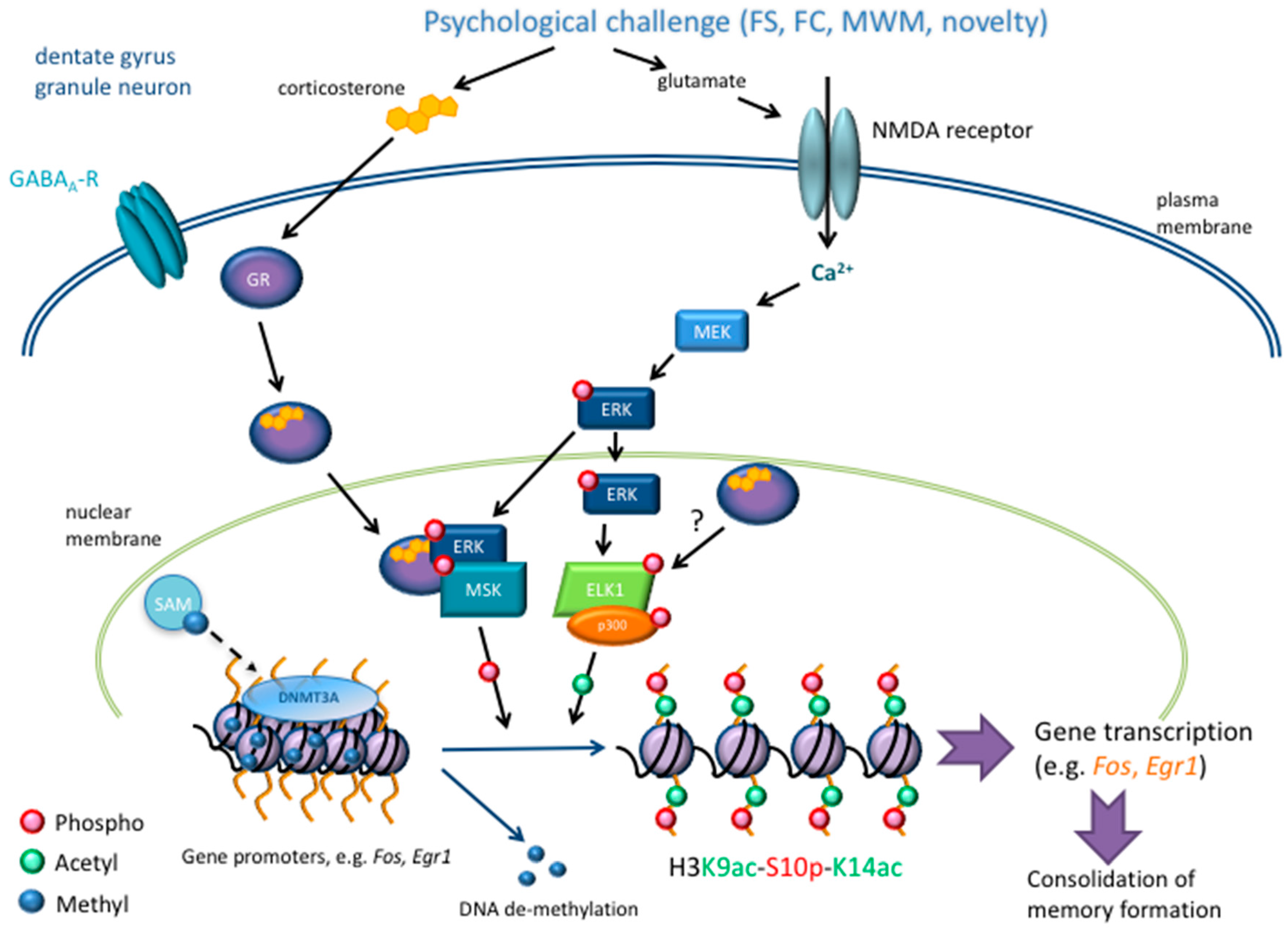{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epigenetics
2.1. Epigenetic Mechanisms in Stress-Related Learning and Memory Paradigms
2.1.1. Acetylation
2.1.2. Phosphorylation
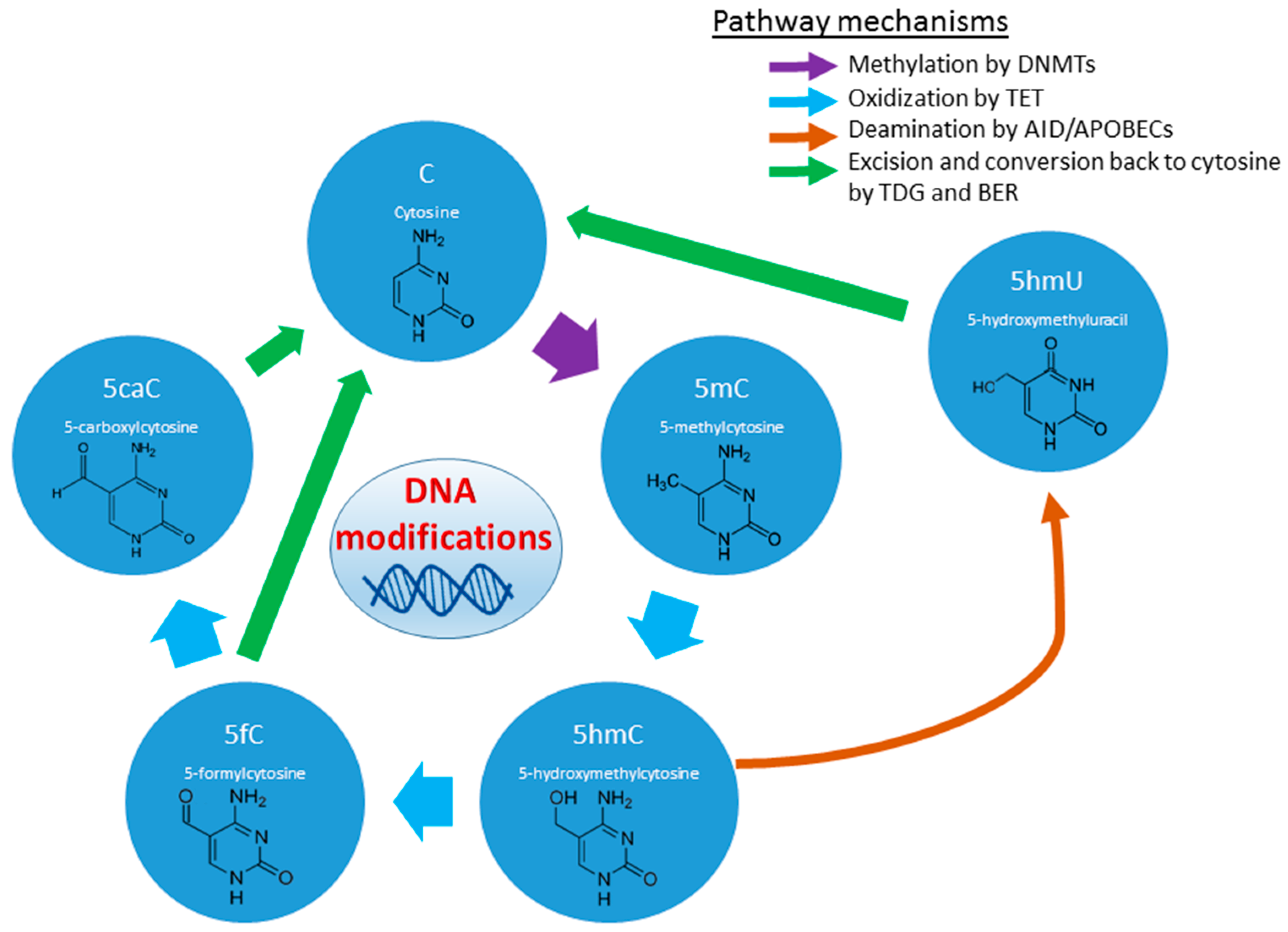
2.1.3. Histone and DNA Methylation
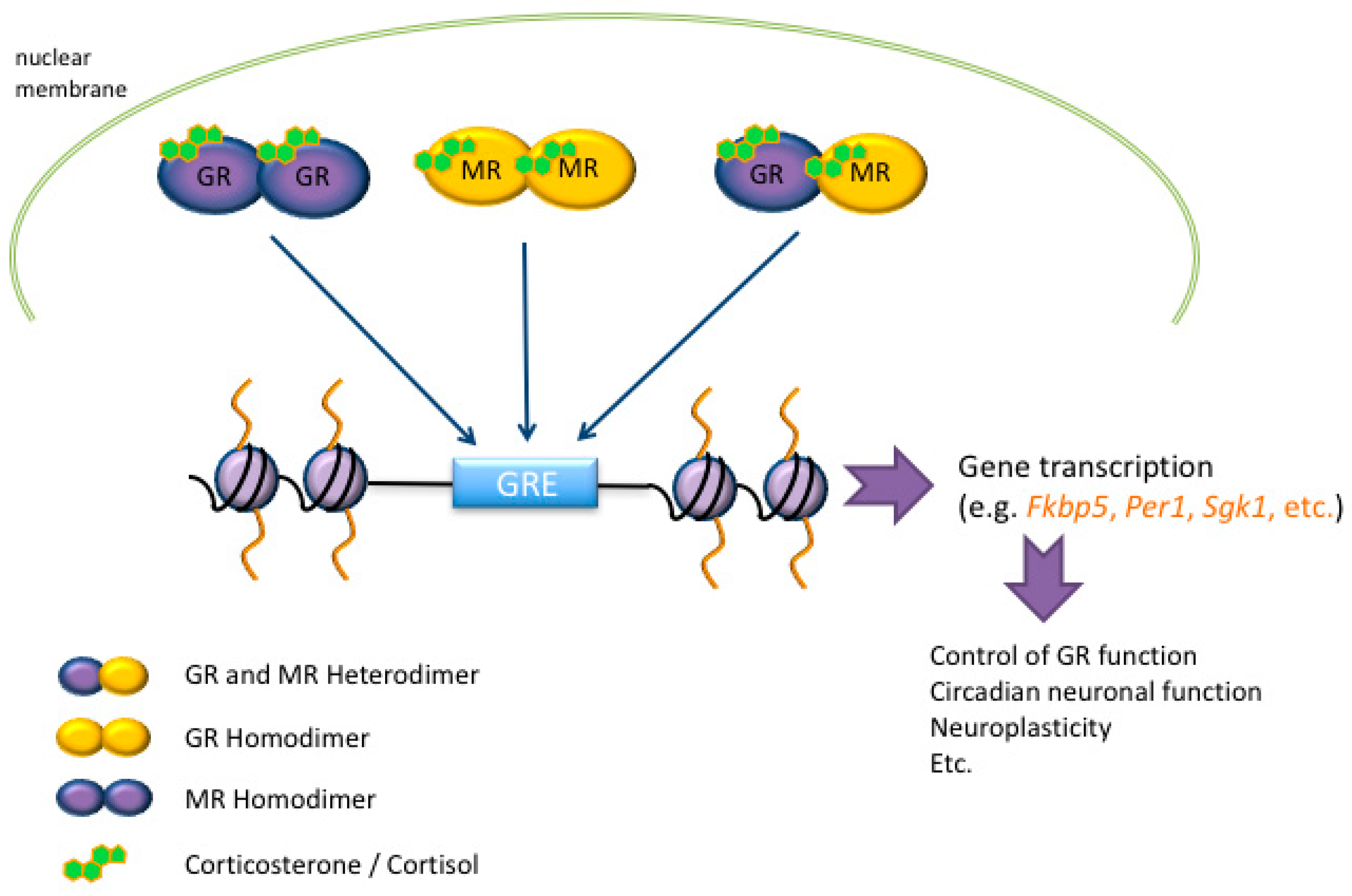
3. Glucocorticoid Hormone Action at the Genomic Level after Stress
4. Early Life Stress, Epigenetic Dysregulation and Neuropsychiatric Disorders
4.1. Prenatal Exposure
4.2. Post-Natal Exposure
4.3. Transgenerational Epigenetics
5. Epigenetics as Biomarkers and Therapeutic Treatments
6. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chrousos, G. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. The neurobiology of stress: From serendipity to clinical relevance. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M. Making memories of stressful events: A journey along epigenetic, gene transcription, and signaling pathways. Front. Psychiatry 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; De Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; De Kloet, E.R. Anatomical resolution of two types of corticosterone receptor sites in rat brain with in vitro autoradiography and computerized image analysis. J. Steroid Biochem. 1986, 24, 269–272. [Google Scholar] [CrossRef]
- Van Steensel, B.; Van Binnendijk, E.P.; Hornsby, C.D.; Van der Voort, H.T.M.; Krozowski, Z.S.; De Kloet, E.R.; Van Driel, R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 1996, 109, 787–792. [Google Scholar] [PubMed]
- Arriza, J.L.; Evans, R.M. Human Mineralocorticoid Receptor Cloning and Expression. In Adrenal and Hypertension: From Cloning to Clinic; Raven: Nashwille, TN, USA, 1989; pp. 91–97. [Google Scholar]
- Hollenberg, S.M.; Giguere, V.; Segui, P.; Evans, R.M. Colocalization of DNA-binding and transcriptional activation functions in the human glucocorticoid receptor. Cell 1987, 49, 39–46. [Google Scholar] [CrossRef]
- Mifsud, K.R.; Reul, J.M.H.M. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc. Natl. Acad. Sci. USA 2016, 113, 11336–11341. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joels, M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of non-genomic corticosteroid signalling. Mol. Cell. Endocrinol. 2012, 350, 299–309. [Google Scholar]
- Saunderson, E.; Spiers, H.; Mifsud, K.; Gutierrez-Mecinas, M.; Trollope, A.; Shaikh, A.; Mill, J.; Reul, J.M.H.M. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2016, 113, 4830–4835. [Google Scholar] [CrossRef] [PubMed]
- Gutièrrez-Mecinas, M.; Trollope, A.F.; Collins, A.; Morfett, H.; Hesketh, S.; Kersanté, F.; Reul, J.M.H.M. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 13806–13811. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Endeavour 1. Epigenotpye 1942, 1, 18–20. [Google Scholar]
- Lester, B.M.; Tronick, E.; Nestler, E.; Abel, T.; Kosofsky, B.; Kuzawa, C.W.; Marsit, C.J.; Maze, I.; Meaney, M.J.; Monteggia, L.M.; et al. Behavioral Epigenetics. Ann. N. Y. Acad. Sci. 2011, 1226, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Agranoff, B.W.; Davis, R.E.; Casola, L.; Lim, R. Actinomycin D blocks formation of memory of shock-avoidance in goldfish. Science 1967, 158, 1600–1601. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; Collins, A.; Saliba, R.S.; Mifsud, K.R.; Carter, S.D.; Gutierrez-Mecinas, M.; Qian, X.; Linthorst, A.C. Glucocorticoids, epigenetic control and stress resilience. Neurobiol. Stress 2014, 15, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [PubMed]
- Bousiges, O.; Vasconcelos, A.P.; Neidl, R.; Cosquer, B.; Herbeaux, K.; Panteleeva, I.; Loeffler, J.P.; Cassel, J.C.; Boutillier, A.L. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology 2010, 35, 2521–2537. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.; Fletcher, B.; Kelley-Bell, B.; Kim, D.; Gallagher, M.; Rapp, P. Age-related memory impairment is associated with disrupted multivariate epigenetic coordination in the hippocampus. PLoS ONE 2012, 7, e33249. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Mifsud, K.R.; Reul, J.M.H.M. Distinct epigenetic and gene expression changes in rat hippocampal neurons after Morris water maze training. Front. Behav. Neurosci. 2015, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; de Cock, S.; Schild, V.; Veldhuis, H.D. Antiglucocorticoid RU38486 attenuates retention of a behaviour and disinhibits the hypothalamic-pituitary-adrenal axis at different brain sites. Neuroendocrinology 1988, 47, 109–115. [Google Scholar]
- Bachmann, C.G.; Bilang-Bleuel, A.; De Carli, S.; Linthorst, A.C.E.; Reul, J.M.H.M. The selective glucocorticoid receptor antagonist ORG 34116 decreases immobility time in the forced swim test and affects CREB phosphorylation in rat brain. Neuroendocrinology 2005, 81, 129–136. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Molendijk, M.L. Coping with the Forced Swim Stressor: Towards Understanding an Adaptive Mechanism. Neural Plast. 2016, 2016, 6503162. [Google Scholar] [CrossRef] [PubMed]
- Bilang-Bleuel, A.; Ulbricht, S.; Chandramohan, Y.; De Carli, S.; Droste, S.K.; Reul, J.M.H.M. Psychological stress increases histone H3 phosphorylation in adult dentate gyrus granule neurons: Involvement in a glucocorticoid receptor-dependent behavioural response. Eur. J. Neurosci. 2005, 22, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, Y.; Droste, S.K.; Arthur, J.S.; Reul, J.M.H.M. The forced swimming-induced behavioural immobility response involves histone H3 phospho-acetylation and c-Fos induction in dentate gyrus granule neurons via activation of the N-methyl-d-aspartate/extracellular signal-regulated kinase/mitogen- and stress-activated kinase signalling pathway. Eur. J. Neurosci. 2008, 27, 2701–2713. [Google Scholar] [PubMed]
- Chandramohan, Y.; Droste, S.K.; Reul, J.M.H.M. Novelty stress induces phospho-acetylation of histone H3 in rat dentate gyrus granule neurons through coincident signalling via the N-methyl-d-aspartate receptor and the glucocorticoid receptor: Relevance for c-fos induction. J. Neurochem. 2007, 101, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S. MSK activation and physiological roles. Front. Biosci. 2008, 13, 5866–5879. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Vickers, E.; Brehm, A.; Kouzarides, T.; Sharrocks, A.D. Temporal recruitment of the mSin3A-histone deacetylase corepressor complex to the ETS domain transcription factor Elk-1. Mol. Cell. Biol. 2001, 21, 2802–2814. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Yang, S.H.; Maeda, Y.; Sladek, F.M.; Sharrocks, A.D.; Martins-Green, M. MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the co-activator p300. EMBO J. 2003, 22, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Vanhoutte, P.; Pagès, C.; Caboche, J.; Laroche, S. The MAPK/ERK Cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentation-dependent gene expression in the dentate gyrus in vivo. J. Neurosci. 2000, 20, 4563–4572. [Google Scholar] [PubMed]
- Reul, J.M.H.M.; Chandramohan, Y. Epigenetic mechanisms in stress-related memory formation. Psychoneuroendocrinology 2007, 32 (Suppl. S1), S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.L.; Rose, S.; Barratt, M.J.; Mahadevan, L.C. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19, 3714–3726. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.W.; Errington, M.L.; French, P.J.; Fine, A.; Bliss, T.V.; Garel, S.; Charnay, P.; Bozon, B.; Laroche, S.; Davis, S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 2001, 4, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; Nutt, D.J. Glutamate and cortisol—A critical confluence in PTSD? J. Psychopharmacol. 2008, 22, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Selcher, J.C.; Atkins, C.M.; Trzaskos, J.M.; Paylor, R.; Sweatt, J.D. A necessity for MAP kinase activation in mammalian spatial learning. Learn. Mem. 1999, 6, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S.; Tsien, J.Z.; McHugh, T.J.; Huerta, P.; Blum, K.I.; Wilson, M.A. Hippocampal CA1-region-restricted knockout of NMDAR1 gene disrupts synaptic plasticity, place fields, and spatial learning. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 225–238. [Google Scholar] [PubMed]
- Chwang, W.B.; Arthur, J.S.; Schumacher, A.; Sweatt, J.D. The Nuclear Kinase Mitogen- and Stress-Activated Protein Kinase 1 Regulates Hippocampal Chromatin Remodeling in Memory Formation. J. Neurosci. 2007, 27, 12732–12742. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone methylation regulates memory formation. J. Neurosci. 2010, 30, 3589–3599. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [PubMed]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [PubMed]
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.; Carmouche, R.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wang, K.Y.; Shen, C.K. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J. Biol. Chem. 2012, 287, 33116–33121. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, K.; Shen, C. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013, 288, 9084–9091. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.; Manashirov, S.; Zwang, R.; Gil, S.; Tsoory, M.; Shemesh, Y.; Chen, A. Dnmt3a in the Medial Prefrontal Cortex Regulates Anxiety-Like Behavior in Adult Mice. J. Neurosci. 2016, 36, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; McGonagle, K.A.; Zhao, S.; Nelson, C.B.; Hughes, M.; Eshleman, S.; Wittchen, H.U.; Kendler, K.S. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch. Gen. Psychiatry 1994, 51, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.A.; Neighbors, H.W.; Griffith, D.M. The experience of symptoms of depression in men vs women: Analysis of the National Comorbidity Survey Replication. JAMA Psychiatry 2013, 70, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.; Pfau, M.; Purushothaman, I.; Ahn, H.; Golden, S.A.; Christoffel, D.J.; Magida, J.; Brancato, A.; Takahashi, A.; Flanigan, M.E.; et al. Sex Differences in Nucleus Accumbens Transcriptome Profiles Associated with Susceptibility versus Resilience to Subchronic Variable Stress. J. Neurosci. 2015, 35, 16362–16376. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Christian, K.; He, C.; Jin, P.; Ming, G.L.; Song, H. Epigenetic mechanisms in neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Li.; Wei, W.; Zhao, Q.; Dai, C.; Leighton, L.; Bredy, T.W. Discovery of a New Mechanism of ING1 Regulating Extinction Memory in Mice. 2017. Available online: https://ibangsmadrid2017.sched.com/event/AQd6/outstanding-travel-award-talks (accessed on 5 June 2017).
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Soulé, J.; Messaoudi, E.; Bramham, C.R. Brain-derived neurotrophic factor and control of synaptic consolidation in the adult brain. Biochem. Soc. Trans. 2006, 34, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Kangaspeska, S.; Stride, B.; Métivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, K.R.; Saunderson, E.A.; Spiers, H.; Carter, S.D.; Trollope, A.F.; Mill, J.; Reul, J.M.H.M. Rapid Down-Regulation of Glucocorticoid Receptor Gene Expression in the Dentate Gyrus after Acute Stress in vivo: Role of DNA Methylation and MicroRNA Activity. Neuroendocrinology 2017, 104, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Vreugdenhil, E.; Verissimo, C.S.; Mariman, R.; Kamphorst, J.T.; Barbosa, J.S.; Zweers, T.; Champagne, D.L.; Schouten, T.; Meijer, O.C.; De Kloet, E.R.; et al. MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: Implications for glucocorticoid responsiveness in the brain. Endocrinology 2009, 150, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Reul, J.M.H.M. Feedback action and tonic influence of corticosteroids on brain function: A concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology 1987, 12, 83–105. [Google Scholar] [CrossRef]
- Revollo, J.R.; Cidlowski, J.A. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann. N. Y. Acad. Sci. 2009, 1179, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Beato, M. Gene regulation by steroid hormones. Cell 1989, 56, 335–344. [Google Scholar] [CrossRef]
- Jenkins, B.D.; Pullen, C.B.; Darimont, B.D. Novel glucocorticoid receptor coactivator effector mechanisms. Trends Endocrinol. Metab. 2001, 12, 122–126. [Google Scholar] [CrossRef]
- Rogatsky, I.; Ivashkiv, L.B. Glucocorticoid modulation of cytokine signaling. Tissue Antigens 2006, 68, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; Van den Bosch, F.R.; De Kloet, E.R. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: Functional implications. J. Endocrinol. 1987, 115, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Gesing, A.; Bilang-Bleuel, A.; Droste, S.K.; Linthorst, A.C.E.; Holsboer, F.; Reul, J.M.H.M. Psychological stress increases hippocampal mineralocorticoid receptor levels: Involvement of corticotropin-releasing hormone. J. Neurosci. 2001, 21, 4822–4829. [Google Scholar] [PubMed]
- Linthorst, A.C.E.; Reul, J.M.H.M. Stress and the brain: Solving the puzzle using microdialysis. Pharmacol. Biochem. Behav. 2008, 90, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Droste, S.K.; de Groote, L.; Atkinson, H.C.; Lightman, S.L.; Reul, J.M.H.M.; Linthorst, A.C.E. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology 2008, 149, 3244–3253. [Google Scholar] [CrossRef] [PubMed]
- Droste, S.K.; Collins, A.; Lightman, S.L.; Linthorst, A.C.E.; Reul, J.M.H.M. Distinct, time-dependent effects of voluntary exercise on circadian and ultradian rhythms and stress responses of free corticosterone in the rat hippocampus. Endocrinology 2009, 150, 4170–4179. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Droste, S.; Lightman, S.; Reul, J.M.H.M.; Linthorst, A. Circadian and ultradian rhythms of free glucocorticoid hormone are highly synchronized between the blood, the subcutaneous tissue, and the brain. Endocrinology 2012, 153, 4346–4353. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.M.H.M.; Gesing, A.; Droste, S.K.; Stec, I.S.M.; Weber, A.; Bachmann, C.G.; Bilang-Bleuel, A.; Holsboer, F.; Linthorst, A.C.E. The brain mineralocorticoid receptor:Greedy for ligand, mysterious in function. Eur. J. Pharmacol. 2000, 405, 235–249. [Google Scholar] [CrossRef]
- Polman, J.; de Kloet, E.; Datson, N. Two populations of glucocorticoid receptor-binding sites in the male rat hippocampal genome. Endocrinology 2013, 154, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Van Weert, L.T.; Buurstede, J.C.; Mahfouz, A.; Braakhuis, P.S.; Polman, J.A.; Sips, H.C.; Roozendaal, B.; Balog, J.; de Kloet, E.R.; Datson, N.A.; et al. NeuroD factors discriminate mineralocorticoid from glucocorticoid receptor DNA binding in the male rat brain. Endocrinology 2017, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Trapp, T.; Rupprecht, R.; Castren, M.; Reul, J.M.H.M.; Holsboer, F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: A new principle of glucocorticoid action in the CNS. Neuron 1994, 13, 1457–1462. [Google Scholar] [CrossRef]
- Hubler, T.R.; Scammell, J.G. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 2004, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Nemeroff, C.B. The influence of childhood disciplinary experience on the development of alcoholism and depression. J. Child Psychol. Psychiatry 2001, 28, 399–415. [Google Scholar]
- Enoch, M. The role of early life stress as a predictor for alcohol and drug dependence. Psychopharmacology 2011, 214, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S.; et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; O’Donovan, M.C.; Thapar, A.; Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 2011, 198, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Engel, S.M.; Brand, S.R.; Seckl, J.; Marcus, S.M.; Berkowitz, G.S. Transgenerational effects of posttraumatic stress disorder in babies of mothers exposed to the World Trade Center attacks during pregnancy. J. Clin. Endocrinol. Metab. 2005, 90, 4115–4118. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Halligan, S.L.; Bierer, L.M. Cortisol levels in adult offspring of Holocaust survivors: Relation to PTSD symptom severity in the parent and child. Psychoneuroendocrinology 2002, 27, 171–180. [Google Scholar] [CrossRef]
- Meewisse, M.L.; Reitsma, J.B.; de Vries, G.J.; Gersons, B.P.; Olff, M. Cortisol and post-traumatic stress disorder in adults: Systematic review and meta-analysis. Br. J. Psychiatry 2007, 191, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Teicher, M.H.; Seckl, J.R.; Grossman, R.A.; Morris, A.; Bierer, L.M. Parental posttraumatic stress disorder as a vulnerability factor for low cortisol trait in offspring of holocaust survivors. Arch. Gen. Psychiatry 2007, 64, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Lehrner, A.; Bierer, L.M.; Passarelli, V.; Pratchett, L.C.; Flory, J.D.; Bader, H.; Harris, I.R.; Bedi, A.; Daskalakis, N.P.; Makotkine, L.; et al. Maternal PTSD associates with greater glucocorticoid sensitivity in offspring of Holocaust survivors. Psychoneuroendocrinology 2014, 40, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, E.P.; Lumeye, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Hoek, H.W.; Susser, E.; Buck, K.A.; Lumey, L.H.; Lin, S.P.; Gorman, J.M. Schizoid personality disorder after prenatal exposure to famine. Am. J. Psychiatry 1996, 153, 1637–1639. [Google Scholar] [PubMed]
- Xie, L.; Korkmaz, K.S.; Braun, K.; Bock, J. Early life stress-induced histone acetylations correlate with activation of the synaptic plasticity genes Arc and Egr1 in the mouse hippocampus. J. Neurochem. 2013, 125, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Champagne, F.A.; Brown, S.E.; Dymov, S.; Sharma, S.; Meaney, M.J.; Szyf, M. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: Altering epigenetic marking later in life. J. Neurosci. 2005, 25, 11045–11054. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Dias, B.G.; Ressler, K.J. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 2014, 17, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Dietmann, S.; Irie, N.; Leitch, H.G.; Floros, V.I.; Bradshaw, C.R.; Hackett, J.A.; Chinnery, P.F.; Surani, M.A. A Unique Gene Regulatory Network Resets the Human Germline Epigenome for Development. Cell 2015, 16, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Dietz, D.M.; Laplant, Q.; Watts, E.L.; Hodes, G.E.; Russo, S.J.; Feng, J.; Oosting, R.S.; Vialou, V.; Nestler, E.J. Paternal transmission of stress-induced pathologies. Biol. Psychiatry 2011, 70, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, A.B.; Morgan, C.P.; Bronson, S.L.; Revello, S.; Bale, T.L. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J. Neurosci. 2013, 33, 9003–9012. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, A.B.; Morgan, C.P.; Leu, N.A.; Bale, T.L. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc. Natl. Acad. Sci. USA 2015, 112, 13699–13704. [Google Scholar] [CrossRef] [PubMed]
- Gapp, K.; Jawaid, A.; Sarkies, P.; Bohacek, J.; Pelczar, P.; Prados, J.; Farinelli, L.; Miska, E.; Mansuy, I.M. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat. Neurosci. 2014, 17, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2015, 68, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Green, E.D.; Guttmacher, A.E.; Guyer, M.S. A vision for the future of genomics research. Nature 2003, 422, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Daudén, T.E. Pharmacogenetics I. Concept, history, objectives and areas of study. Actas Dermosifiliogr. 2006, 97, 623–629. [Google Scholar] [CrossRef]
- Peedicadyl, J. Pharmacoepigenetics and pharmacoepigenomics. Pharmacogenomics 2008, 9, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Baylin, S. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Van Rijnsoever, M.; Elsaleh, H.; Joseph, D.; McCaul, K.; Lacopetta, B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin. Cancer Res. 2003, 9, 2898–2903. [Google Scholar] [PubMed]
- Smith, J.; Freije, D.; Carpten, J.; Grönberg, H.; Xu, J.; Isaacs, S.; Brownstein, M.; Bova, G.; Guo, H.; Bujnovszky, P.; et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science 1996, 274, 1371–1374. [Google Scholar] [CrossRef]
- Belinsky, S. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer 2004, 4, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; De Carvalho, D.; Jones, P. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.; Fernandez, A.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.; O’Hanlon, T.; Rider, L.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, L.M.; Natarajan, R. The role of epigenetics in the pathology of diabetic complications. Am. J. Physiol. Renal Physiol. 2010, 299, F14–F25. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.; Ito, K.; Barnes, P. Histone deacetylation: An important mechanism in inflammatory lung diseases. COPD J. Chron. Obstruct. Pulm. Dis. 2005, 2, 445–455. [Google Scholar] [CrossRef]
- Urdinguio, R.; Sanchez-Mut, J.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trollope, A.F.; Mifsud, K.R.; Saunderson, E.A.; Reul, J.M.H.M. Molecular and Epigenetic Mechanisms Underlying Cognitive and Adaptive Responses to Stress. Epigenomes 2017, 1, 17. https://doi.org/10.3390/epigenomes1030017
Trollope AF, Mifsud KR, Saunderson EA, Reul JMHM. Molecular and Epigenetic Mechanisms Underlying Cognitive and Adaptive Responses to Stress. Epigenomes. 2017; 1(3):17. https://doi.org/10.3390/epigenomes1030017
Chicago/Turabian StyleTrollope, Alexandra F., Karen R. Mifsud, Emily A. Saunderson, and Johannes M. H. M. Reul. 2017. "Molecular and Epigenetic Mechanisms Underlying Cognitive and Adaptive Responses to Stress" Epigenomes 1, no. 3: 17. https://doi.org/10.3390/epigenomes1030017
APA StyleTrollope, A. F., Mifsud, K. R., Saunderson, E. A., & Reul, J. M. H. M. (2017). Molecular and Epigenetic Mechanisms Underlying Cognitive and Adaptive Responses to Stress. Epigenomes, 1(3), 17. https://doi.org/10.3390/epigenomes1030017