Abstract
The gene EHMT1 that encodes the Euchromatic Histone Methyltransferase-1, also known as GLP (G9a-like protein), has been associated with a number of neurodevelopmental and neurodegenerative disorders. GLP is a member of the euchromatic lysine histone methyltransferase family, along with EHMT2 or G9A. As its name implies, Ehmt1/GLP is involved in the addition of methyl groups to histone H3 lysine 9, a generally repressive mark linked to classical epigenetic process such as genomic imprinting, X-inactivation, and heterochromatin formation. However, GLP also plays both a direct and indirect role in regulating DNA-methylation. Here, we discuss what is currently known about the biochemical function of Ehmt1/GLP and its association, via various genetic studies, with brain disorders.
1. Introduction
Euchromatic Histone Methyltransferase-1 (EHMT1), also known as GLP (G9a-like protein), is a member of the euchromatic lysine histone methyltransferase family, along with EHMT2 or G9A [1,2,3]. These two proteins are part of a superfamily of lysine methyltransferases that are characterised by the presence of a SET domain. Within this superfamily GLP and G9A are a part of the Suv39h1 family of lysine methyltransferases responsible for the mono-, di-, and tri-methylation at lysine residues [4]. The SET/pre-SET domains have been identified as key for methyltransferase activity [5]. These proteins, and their methyltransferase activity, are highly conserved through evolution with a large number of orthologues having been characterised in other species, including EHMT in Drosophila melanogaster, encoded by a single gene that appears to share functional similarities with both GLP and G9a [6].
Both GLP and G9A also uniquely contain ankyrin repeats in their protein structure. These ankyrin repeats, although unique within the methyltransferase family of proteins, are also found in a wide range of proteins with differing functions including cell adhesion and synaptic functioning [7,8]. Ankyrin repeats are believed to be important for protein-protein interactions, suggesting a similar role GLP and G9a [9]. The Ank domains in GLP and G9a have been shown to preferentially bind to Histone 3 (H3) tails at mono- and di-methylated K9 [10], providing evidence of the proteins being able to recognise the marks.
Although widely recognised as histone methyltransferases, there is growing evidence of the SET domain superfamily of enzymes interacting with other proteins. GLP and G9a have both been identified to methylate non-histone target proteins such as WIZ, a zinc finger protein with which GLP/G9a form a stable repressive complex [11]; p53, a tumour suppressor [12]; and CDYL, a RE1-Silencing Transcription factor (REST) co-repressor [11]. GLP has also been found to target DNA methyltransferases, DNMT1 and DNMT3/3a [13]. For the most part, the greater roles of these interactions are not understood.
2. Methyltransferase Activity
GLP mediates the addition of mono- and di- methyl groups to the lysine 9 (K9) position on histone H3 within euchromatic regions of the genome, doing so via its catalytic SET/pre-SET domains [1,2]. This is generally associated with repression of transcription, and is associated with classical epigenetic mechanisms of transcriptional silence such as X-inactivation [14], genomic imprinting [15,16], and globally within regions of heterochromatin [17]. GLP is often found within repressive complexes, and notably forms a stoichiometric complex with G9a that appears to be vital for in vivo dimethylation at H3K9 [2]. GLP is also a member of a large complex that includes the Neuron-Restrictive Silencer Factor (NRSF/REST) repressive unit important for repressing neuronal genes in progenitors [18,19]. Finally, GLP has been shown to also form a mega complex with G9a, SETDB1, and Suv39h1 that, in turn, appears to regulate G9a target genes [20].
Despite apparent redundancy in the overlapping function of GLP and G9a, Tachibana et al. (2005) [2] showed that knocking out either protein leads to significantly reduced H3K9 dimethylation, with no further reduction being seen in a double knockout, suggesting a lack of compensation of function by either GLP or G9A. Furthermore, homozygous deletion of GLP leads to embryonic lethality in mice, suggesting a critical role during development that cannot be compensated for by the G9a alone. This central importance for a role in development is complimented with evidence of GLP repressing E2F/MYC-responsive genes important for cell cycle regulation [21] and its aforementioned role in REST complex [19].
3. DNA Methylation
There is growing evidence for the involvement of GLP and the repressive chromatin mark H3K9me2 in the re-establishment and maintenance of DNA methylation. DNA methylation needs to be re-established upon the formation of the hemi-methylated daughter strands after DNA replication. A reduction of either GLP or G9A leads to a decrease in DNA methylation [22], and H3K9me2 marks appear necessary for the maintenance process [23,24]. There is some evidence of DNMT1 interacting with G9a, regulating H3K9 methylation in the HCT116 cancerous colon immortalised cells, where knocking out DNMT1 lead to a reduction in H3K9me2 [25]. Low DNA methylation in embryonic stem cells (ESCs) cultured in 2i serum was associated with decreased DNA methylation maintenance and associated protein E3 ubiquitin-protein ligase (UHRF1) and decreased GLP/G9a expression, coupled with an upregulation of several H3K9 demethylases [26]. However, the exact mechanism by which GLP mediates the maintenance of DNA methylation is not fully understood. UHRF1 is known to co-localise and directly interact with DNMT1 and aids in the maintenance of the methylation. Through its SET/RING domain it binds preferentially to hemi-methylated DNA, recruiting DNMT1 to re-establish methylation [27,28]. UHRF1 was also shown to bind to H3K9me2/3 through its tandem Tudor domain and appears to bind DNMT1 to mediate DNA methylation maintenance in an H3K9me2/3 dependent manner [27]. A recent paper however has increased the current understanding of this process; DNA ligase 1 (LIG1), a novel target of GLP is methylated on its H3K9 mimic, UHRF1 then binds to the mimic, and in turn this binding leads to the recruitment of UHRF1 to hemi-methylated sites, mediating its activity [29] (Figure 1). UHRF1 was also found to more readily bind to the LIG1 mimic compared to H3K9me2/3. Current evidence is suggestive of complimentary role of GLP in histone and DNA methylation.
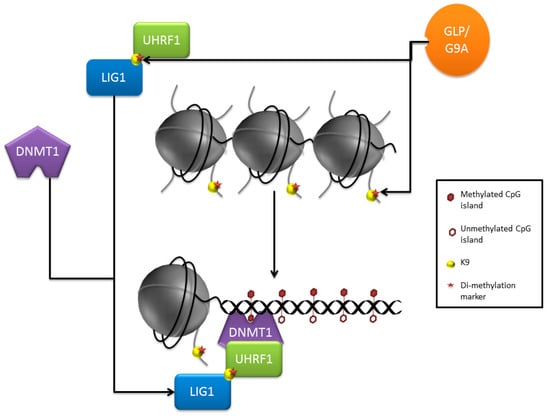
Figure 1.
Maintenance of DNA methylation: Glp and G9a are known to target H3K9 for mono and di-methylation, they have also recently been found to methylate the H3K9 mimic on DNA ligase 1 (LIG1). UHRF1 can recognise and bind both H3K9me2/3 marks as well as the methylated mimic on LIG1. UHRF1 binds to methylated LIG1 with a high affinity, this in turn mediates its activity to recognise and preferentially bind to hemi-methylated DNA strands, recruiting DNA methyltransferase 1 (DNMT1) to re-establish methylation and CpG islands.
4. GLP in Brain Disorder
In humans, the gene encoding GLP, EHMT1, is found on the long arm of chromosome 9, specifically at q34.3. A range of genetic studies has linked loss (haploinsufficiency) or mutation of one copy of this gene with a number of brain disorders, particularly neurodevelopmental disorders. These data add to the growing image of neurodevelopmental disorders being, in part, “epigeneopathies”, as GLP and various other important enzymes in histone modification have been implicated in the pathogenesis of neurodevelopmental disorders (reviewed in [30]).
4.1. Intellectual Disability
Intellectual disability affects approximately 1–3% of the population and is characterised by reduced cognitive ability and some adaptive behaviour problems [31]. Intellectual disability can be sporadic and isolated, or as an identifying phenotype in a complex syndrome and is a key symptom in various syndromes including Down syndrome and fragile X syndrome [30].
GLP is most notably associated with Kleefstra syndrome, an intellectual disability multi-system syndrome associated with congenital heart defects, hypotonia, and dysmorphisms [32]. Kleefstra syndrome is caused by the haploinsufficiency of GLP, either through single mutations or 9q34.3 microdeletions [32,33]. More recently, a sub-group of patients suffering from Kleefstra syndrome that do not have the deletion or mutation of GLP have been identified. Interestingly however, whilst GLP appears genetically intact in this sub-group, individuals do carry mutations in other genes known to directly interact with GLP, such as MBD5, another epigenetic regulator [34]. This has helped the development of the central role of GLP in the pathogenesis of Kleefstra syndrome, and its larger impact in epigenetic regulation.
Outside of Kleefstra syndrome, de novo deletions of GLP have been detected in severe intellectual disability Copy Number Variant (CNV studies) [35,36,37], and GLP associated chromatin regulators have also been linked to sporadic intellectual disability in chromosomal microarray [37]. Furthermore, exome studies of patients with intellectual disability again link GLP to a wider epigenetic regulatory network important to cognitive function [31,38]. Recently, exome sequencing of approximately 4300 families with members suffering from developmental disorders identified GLP as a genome-wide significant candidate in the pathogenesis of developmental disorders generally [39]. Finally, GLP has also been identified as a pathogenic CNV in patients with intellectual disability and early onset epilepsy [40]. Epilepsy is often associated with ID and developmental delay [38,40], and some patients with Kleefstra syndrome are also known to be comorbid with epilepsy [41].
4.2. Autistic Spectrum Disorders
GLP has also been associated with autism spectrum disorders (ASDs). ASDs share a common base of phenotypes including dysfunctional social behaviour and restricted adaptive and repetitive behaviours [42]. Interestingly, ASD is also highly co-diagnosed in Kleefstra syndrome patients [43] and is known to be comorbid with a range of other developmental disorders and intellectual disability [44]. CNV analysis of ASD probands identified GLP as de novo mutation [45]. This evidence is strengthened by a recent study where BCA (balanced chromosomal abnormalities) sequencing of 22 autism patients identified GLP microdeletions as a risk in the development of ASD [46]. Finally, exon sequencing of Japanese autism patients also identified two novel rare missense GLP and G9a variants. G9a has also been shown to be elevated in the blood of these ASD patients, suggesting an increasingly restrictive chromatin in the pathogenesis of ASD [47]. These studies link GLP to the overlapping phenotypes seen in intellectual disability disorders and autism.
4.3. Schizophrenia and Psychosis
Schizophrenia affects approximately 1% of the population. This highly heterogeneous diagnosis is regarded to share a genetic basis with various other developmental and psychiatric disorders, particularly autism spectrum disorders [48,49]. A population based study showed an overlapping co-diagnosis of schizophrenia and intellectual disability, with over 30% of patients diagnosed with intellectual disability being co-diagnosed with a psychiatric illness, of which schizophrenia was overrepresented [50]. Recently, a copy number variation analysis study [51] identified two de novo GLP CNVs as pathogenic variants in schizophrenia, linking GLP to the pathogenesis of the adult onset psychiatric disorder. Interestingly, diagnosis of schizophrenia was associated with increased GLP and H3K9me2 in post-mortem brain samples, and increased GLP expression was also linked to worsening symptoms [52]. Increased GLP and H3K9me2 marks, and thus a restrictive chromatin state, as seen in ASD, could possibly therefore be considered a marker for schizophrenia, as well as a marker for potential prognosis.
Interestingly, there is now growing evidence that Kleefstra syndrome patients have developed regressive phenotype, and develop adult onset psychosis [53,54]. Therefore, either maintained reduction of GLP’s function, or an early developmental trigger due to GLP haploinsufficiency leads to psychosis in adulthood. Evidence presented here shows that both an increase and decrease in GLP and H3K9me2 appears to lead to similar phenotypes in psychosis, pointing towards GLP’s importance in maintaining the homeostasis of the epigenome, and changes in either direction would lead to impairment.
4.4. Evidence of H3K9me2 in Neurodegeneration
H3K9me2, a marker of heterochromatin, initiated by G9A and GLP has been associated with neurodegeneration in human and animal models. H3K9 methylation is found to increase with age, and is linked to cognitive decline. Perhaps as a further link with pathological aging, H3K9 methylation was increased in the brains of 3xTg-AD mouse model of Alzheimer’s disease [55]. Conversely however, in post-mortem brains of Alzheimer disease patients H3K9me2 was in fact reduced, this was complimented by reduced H3K9me2 in Tau neurodegenerative Drosophila and mouse models, where there is evidence of global loss of heterochromatin [56]. α-Synuclein, a protein associated with Parkinson’s disease and other neurodegenerative disorders, was found to increase the levels of H3K9me2, and overexpression of the protein would lead to an increasingly restrictive chromatin in Parkinson’s disease [57]. H3K9me2 has also been associated with Huntington’s disease (HD), where the marker is shown to be in the striatum of HD patients [58]. The growing evidence of H3K9me2 in the pathogenesis of neurodegenerative disorders is of notable interest due to the previously mentioned regressive phenotype in Kleefstra syndrome patients, with patients showing increased severity in behavioural and motor deficiencies, developing apathy, and also showing signs of subcortical abnormalities [53], suggesting a neurodegenerative course in Kleefstra syndrome prognosis.
5. Conclusions
EHMT1/GLP can be regarded as a key regulator in neurodevelopment, and thus has been widely identified in neurodevelopmental and, to a lesser extent, neurodegenerative disorders. Interestingly, the neurodevelopmental disorders in which EHMT1/GLP has been associated are known to share a molecular pathogenesis, with many of them often being comorbid in their nature. GLP, due to its dynamic function, has been identified in these studies as a cross diagnostic risk factor. From microdeletions to missense variations and single mutations, it would appear GLP has a pleiotropic effect on development and developmental disorders: from early autistic phenotypes to a more regressive phenotype and development of psychosis in the adult, as seen in Kleefstra patients. It may very likely continue on to lead to a more degenerative phenotype, with various neurodegenerative disorders showing a pathogenesis linked to continued imbalance in H3K9me2 markers.
Examining the role epigenetics as a whole on the pathogenesis of neurodevelopmental disorders, with GLP as an example, will lead to a better understanding of their pathogenesis that will likely help inform the involvement of other non-epigenetic genes to lead away from narrow pathogenic tracks such as ‘synaptopathies’ to a more elaborate framework of developmental genes and environment. The understanding of this interplay is of high importance for novel and improved treatments of brain disorders, either by the direct targeting of epigenetic regulators or through targeting upstream and downstream mechanisms identified through basic studies of their modes of action.
Author Contributions
M.A.A. and A.R.I. wrote the paper.
Conflicts of Interest
The authors state they do not have any interests that may conflict with the topic of this article.
References
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Trievel, R.C.; Beach, B.M.; Dirk, L.M.; Houtz, R.L.; Hurley, J.H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 2002, 111, 91–103. [Google Scholar] [CrossRef]
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.M.; Kochinke, K.; Oortveld, M.A.; Marks, H.; Kramer, D.; de Jong, E.K.; Asztalos, Z.; Westwood, J.T.; Stunnenberg, H.G.; Sokolowski, M.B.; et al. Epigenetic regulation of learning and memory by Drosophila EHMT/G9a. PLoS Biol. 2011, 9, e1000569. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, Y.; Wu, C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J. Cell Sci. 1999, 112, 4589–4599. [Google Scholar] [PubMed]
- Lim, S.; Sala, C.; Yoon, J.; Park, S.; Kuroda, S.; Sheng, M.; Kim, E. Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Mol. Cell. Neurosci. 2001, 17, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.E.; Northrop, J.P.; Horton, J.R.; Lee, D.Y.; Zhang, X.; Stallcup, M.R.; Cheng, X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008, 15, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Dhayalan, A.; Murakami, M.; Zhang, X.; Tamas, R.; Jurkowska, R.; Komatsu, Y.; Shinkai, Y.; Cheng, X.; Jeltsch, A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat. Chem. Biol. 2008, 4, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Pérez-Burgos, L.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S.L. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Sun, L.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 533. [Google Scholar] [CrossRef] [PubMed]
- Mermoud, J.E.; Popova, B.; Peters, A.H.; Jenuwein, T.; Brockdorff, N. Histone H3 lysine 9 methylation occurs rapidly at the onset of random X chromosome inactivation. Curr. Biol. 2002, 12, 247–251. [Google Scholar] [CrossRef]
- Xin, Z.; Tachibana, M.; Guggiari, M.; Heard, E.; Shinkai, Y.; Wagstaff, J. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 2003, 278, 14996–15000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Termanis, A.; Özkan, B.; Bao, X.X.; Culley, J.; de Lima Alves, F.; Rappsilber, J.; Ramsahoye, B.; Stancheva, I. G9a/GLP Complex Maintains Imprinted DNA Methylation in Embryonic Stem Cells. Cell Rep. 2016, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.; Mermoud, J.E.; O’Carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 2002, 30, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Roopra, A.; Qazi, R.; Schoenike, B.; Daley, T.J.; Morrison, J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell 2004, 14, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, L.; Robin, P.; Mathieu, J.R.; Souidi, M.; Hinaux, H.; Rougeulle, C.; Harel-Bellan, A.; Ameyar-Zazoua, M.; Ait-Si-Ali, S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell 2010, 37, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Ishiguro, K.; Gaubatz, S.; Livingston, D.M.; Nakatani, Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 2002, 296, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.B.; Maksakova, I.A.; Mohn, F.; Leung, D.; Appanah, R.; Lee, S.; Yang, H.W.; Lam, L.L.; Mager, D.L.; Schübeler, D.; et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008, 27, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Q.; Li, P.; Zhao, Q.; Zhang, J.; Li, J.; Koseki, H.; Wong, J. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 2013, 4, 1563. [Google Scholar] [CrossRef] [PubMed]
- West, P.T.; Li, Q.; Ji, L.; Eichten, S.R.; Song, J.; Vaughn, M.W.; Schmitz, R.J.; Springer, N.M. Genomic distribution of H3K9me2 and DNA methylation in a maize genome. PLoS ONE 2014, 9, e105267. [Google Scholar] [CrossRef] [PubMed]
- Estève, P.O.; Chin, H.G.; Smallwood, A.; Feehery, G.R.; Gangisetty, O.; Karpf, A.R.; Carey, M.F.; Pradhan, S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006, 20, 3089–3103. [Google Scholar] [CrossRef]
- Von Meyenn, F.; Iurlaro, M.; Habibi, E.; Liu, N.Q.; Salehzadeh-Yazdi, A.; Santos, F.; Petrini, E.; Milagre, I.; Yu, M.; Xie, Z.; et al. Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Mol. Cell 2016, 62, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S.; Cornett, E.M.; Goldfarb, D.; DaRosa, P.A.; Li, Z.M.; Yan, F.; Dickson, B.M.; Guo, A.H.; Cantu, D.V.; Kaustov, L.; et al. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forné, I.; Pichler, G.; Hörl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP Recruits UHRF1 to Replicating DNA and Regulates DNA Methylation. Mol. Cell 2017, 67, 550.e5–565.e5. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. An epigenetic framework for neurodevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology 2013, 68, 2–82. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Smidt, M.; Banning, M.J.; Oudakker, A.R.; Van Esch, H.; de Brouwer, A.P.; Nillesen, W.; Sistermans, E.A.; Hamel, B.C.; De Bruijn, D.; et al. Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 2005, 42, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Brunner, H.G.; Amiel, J.; Oudakker, A.R.; Nillesen, W.M.; Magee, A.; Geneviève, D.; Cormier-Daire, V.; Van Esch, H.; Fryns, J.P.; et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Kleefstra, T.; Kramer, J.M.; Neveling, K.; Willemsen, M.H.; Koemans, T.S.; Vissers, L.E.; Wissink-Lindhout, W.; Fenckova, M.; Van Den Akker, W.M.R.; Kasri, N.N.; et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 2012, 91, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Tejada, M.I.; Gecz, J.; Shaw, M.; Corbett, M.; Haan, E.; Thompson, E.; Friend, K.; et al. Targeted Next-Generation Sequencing Analysis of 1,000 Individuals with Intellectual Disability. Hum. Mutat. 2015, 36, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Quintela, I.; Eirís, J.; Gómez-Lado, C.; Pérez-Gay, L.; Dacruz, D.; Cruz, R.; Castro-Gago, M.; Míguez, L.; Carracedo, A.; Barros, F. Copy number variation analysis of patients with intellectual disability from North-West Spain. Gene 2017, 626, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lee, I.G.; Jang, W.; Kim, M.; Kim, Y.; Jang, J.H.; Park, J. Diagnostic exome sequencing identifies a heterozygous MBD5 frameshift mutation in a family with intellectual disability and epilepsy. Eur. J. Med. Genet. 2017, 60, 559–564. [Google Scholar] [CrossRef] [PubMed]
- McRae, J.F.; Clayton, S.; Fitzgerald, T.W.; Kaplanis, J.; Prigmore, E.; Rajan, D.; Ambridge, K. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.E.; Rees, E.; Thompson, R.; Mantripragada, K.; Blake, P.; Jones, G.; Morgan, S.; Jose, S.; Mugalaasi, H.; Archer, H.; et al. Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Med. Genet. 2016, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Hadzsiev, K.; Komlosi, K.; Czako, M.; Duga, B.; Szalai, R.; Szabo, A.; Postyeni, E.; Szabo, T.; Kosztolanyi, G.; Melegh, B. Kleefstra syndrome in Hungarian patients: Additional symptoms besides the classic phenotype. Mol. Cytogenet. 2016, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Cook, E.H.; Anderson, G.M.; Rubenstein, J.L.; Greenough, W.T.; Beckel-Mitchener, A.; Courchesne, E.; Boulanger, L.M.; Powell, S.B.; Levitt, P.R.; et al. Autism as a disorder of neural information processing: Directions for research and targets for therapy. Mol. Psychiatry 2004, 9, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, M.; Okamoto, N.; Harada, N.; Nakamura, T.; Yamamori, S.; Fujita, H.; Niikawa, N.; Matsumoto, N. 9q34.3 deletion syndrome in three unrelated children. Am. J. Med. Genet. A 2004, 126A, 278–283. [Google Scholar] [CrossRef] [PubMed]
- La Malfa, G.; Lassi, S.; Bertelli, M.; Salvini, R.; Placidi, G.F. Autism and intellectual disability: A study of prevalence on a sample of the Italian population. J. Intellect. Disabil. Res. 2004, 48, 262–267. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Iwayama, Y.; Maekawa, M.; Toyota, T.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Esaki, K.; Yamada, K.; Iwata, Y.; et al. Exon resequencing of H3K9 methyltransferase complex genes, EHMT1, EHTM2 and WIZ, in Japanese autism subjects. Mol. Autism 2014, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R.; Pallagrosi, M. Autism Spectrum Disorders and Schizophrenia Spectrum Disorders: Excitation/Inhibition Imbalance and Developmental Trajectories. Front. Psychiatry 2017, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Morgan, V.A.; Leonard, H.; Bourke, J.; Jablensky, A. Intellectual disability co-occurring with schizophrenia and other psychiatric illness: Population-based study. Br. J. Psychiatry 2008, 193, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Chase, K.A.; Gavin, D.P.; Guidotti, A.; Sharma, R.P. Histone methylation at H3K9: Evidence for a restrictive epigenome in schizophrenia. Schizophr. Res. 2013, 149, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; Vermeulen, K.; van de Warrenburg, B.P.; Kleefstra, T. Kleefstra syndrome in three adult patients: Further delineation of the behavioral and neurological phenotype shows aspects of a neurodegenerative course. Am. J. Med. Genet. A 2011, 155A, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; Staal, W.G.; Janzing, J.G.; van Bokhoven, H.; Egger, J.I.M.; Kleefstra, T. Sleep Disturbance as a Precursor of Severe Regression in Kleefstra Syndrome Suggests a Need for Firm and Rapid Pharmacological Treatment. Clin. Neuropharmacol. 2017, 40, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.P.; LaFerla, F.M.; Oddo, S.S.; Brewer, G.J. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age (Dordr.) 2013, 35, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Jäckel, S.; Voigt, A.; Wassouf, Z.; Schulze-Hentrich, J.; Kahle, P.J. α-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci. Rep. 2016, 6, 36328. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).