

Trauma-Induced Coagulopathy: A Review of Specific Molecular Mechanisms


Abstract
1. Introduction
2. Methods
3. Epidemiology and Early Detection of TIC
3.1. Conventional Coagulation Tests
3.2. Viscoelastic Hemostatic Assays
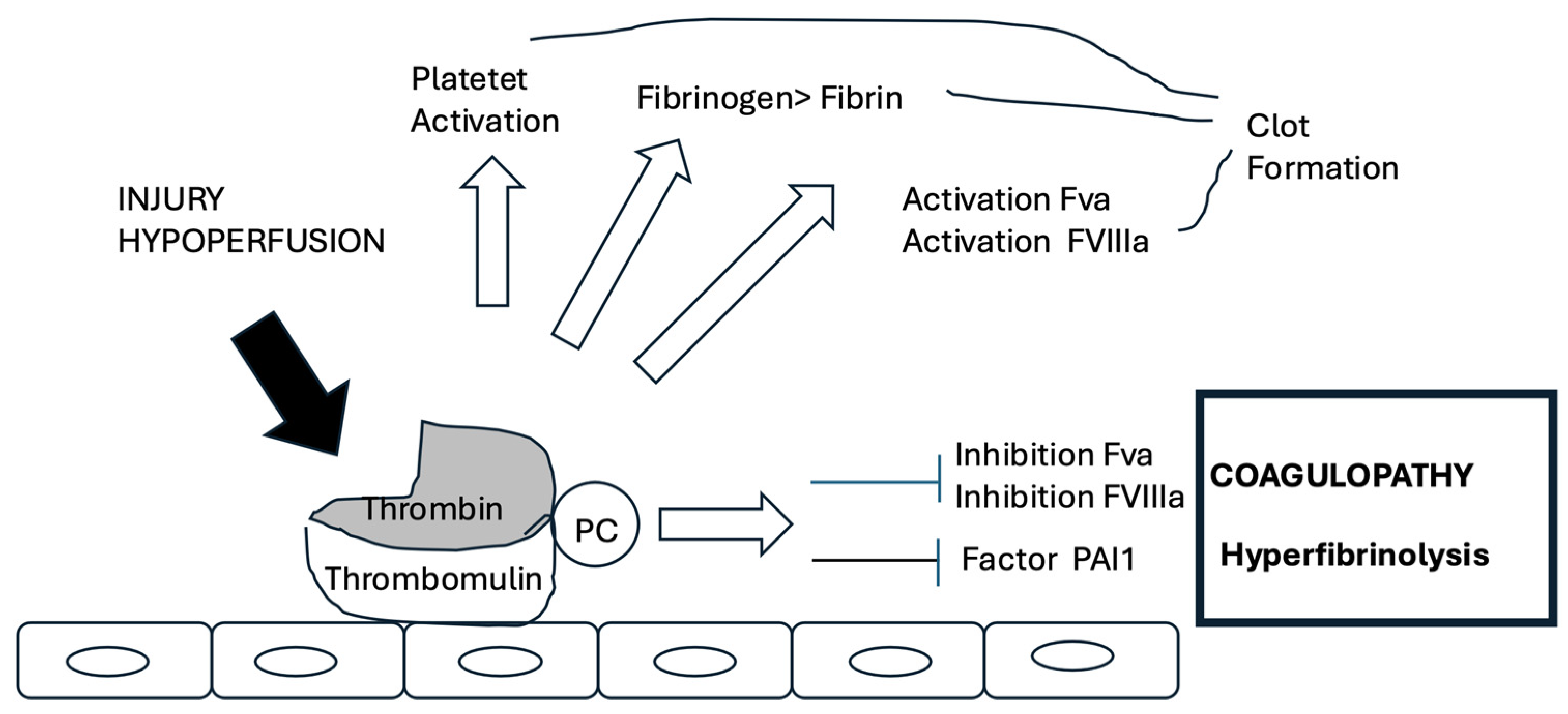
4. Physiopathological Mechanisms
4.1. Lethal Triad: An Outdated Concept
4.2. TIC and DIC: Sibling Coagulopathies
4.3. Hyperfibrinolysis and Fibrinolysis Shutdown
4.3.1. Hyperfibrinolysis
4.3.2. Fibrinolysis Shutdown
4.4. The New Concept: Trauma Endotheliopathy (EoT)
4.4.1. Endothelial Damage
4.4.2. Von Willebrand Factor (VWF) and ADAMTS13
4.4.3. Calcium Influx
4.4.4. RhoA GTPase Activation
4.4.5. Inflammatory Mediators
4.4.6. Extracellular Vesicles (EVs)
4.4.7. Metabolic Dysregulation
5. Therapeutic Evolution in Trauma-Induced Coagulopathy
5.1. Tranexamic Acid: The Cornerstone
5.2. The Goal-Direct Transfusion
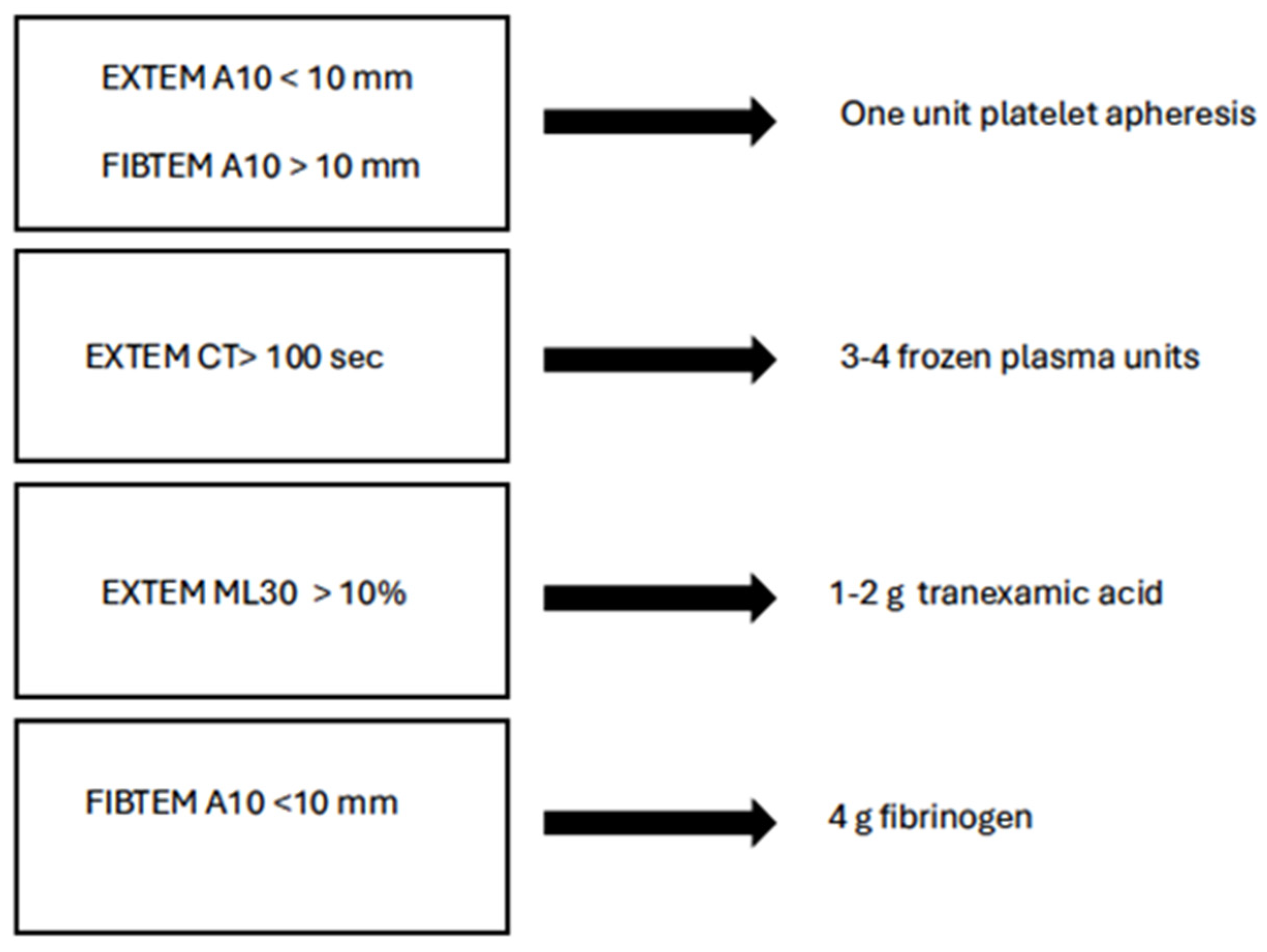
5.3. TEG-Guided Hemostatic Transfusion Strategies
- R-time: A prolonged R-time suggests the requirement for plasma transfusion. For example, an R-time > 4.45 min indicates the need for fresh frozen plasma (FFP) administration to address coagulopathy.
- Angle (α): A decreased angle indicates the need for fibrinogen supplementation. An angle < 67 degrees signals the potential need for fibrinogen concentrates or cryoprecipitate infusion.
- Maximum Amplitude (MA): A reduced MA indicates platelet dysfunction or deficiency. An MA < 60 mm indicates the necessity for platelet transfusion.
- LY30: Elevated LY30 indicates hyperfibrinolysis, which may require antifibrinolytic therapy like tranexamic acid (TXA). An LY30 > 4.55% suggests the potential benefit of antifibrinolytics.
5.4. Improving Trauma Care: Fibrinogen Concentrates
5.5. Pharmacological Interventions
5.6. Calcium Chloride
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma 2003, 54, 1127. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Cardenas, J.C.; Wade, C.E.; Holcomb, J.B. Advances in understanding trauma induced coagulopathy. Blood 2016, 128, 1023–1029. [Google Scholar] [CrossRef]
- Moore, E.E.; Moore, H.B.; Kornblith, L.Z.; Neal, M.D.; Hoffman, M.; Mutch, N.J.; Schöchl, H.; Hunt, B.J.; Sauaia, A. Trauma induced coagulopathy. Nat. Rev. Dis. Primers 2021, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Katrancha, E.D.; Gonzalez, L.S. Trauma-induced coagulopathy. Crit. Care Nurse 2014, 34, 54–63. [Google Scholar] [CrossRef]
- Duque, P.; Calvo, A.; Lockie, C.; Schöchl, H. Pathophysiology of Trauma-Induced Coagulopathy. Transfus. Med. Rev. 2021, 35, 80–86. [Google Scholar] [CrossRef]
- MacLeod, J.B.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early coagulopathy predicts mortality in trauma. J. Trauma 2003, 55, 39–44. [Google Scholar] [CrossRef]
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T.; Simanski, C.; Neugebauer, E.; Bouillon, B.; AG Polytrauma of the German Trauma Society (DGU). Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304. [Google Scholar] [CrossRef]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Matthay, M.A.; Mackersie, R.C.; Pittet, J.F. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812–818. [Google Scholar] [CrossRef]
- Niles, S.E.; McLaughlin, D.F.; Perkins, J.G.; Wade, C.E.; Li, Y.; Spinella, P.C.; Holcomb, J.B. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J. Trauma 2008, 64, 1459–1465. [Google Scholar] [CrossRef]
- Demetriades, D.; Murray, J.; Martin, M.; Velmahos, G.; Salim, A.; Alo, K.; Rhee, P. Pedestrians injured by automobiles: Relationship of age to injury type and severity. J. Am. Coll. Surg. 2004, 199, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.A.; Nelson, T.; McKinley, B.A.; Moore, E.E.; Nathens, A.B.; Rhee, P.; Puyana, J.C.; Beilman, G.J.; Cohn, S.M.; StO2 Study Group. Is there a role for aggressive use of fresh frozen plasma in massive transfusion of civilian trauma patients? Am. J. Surg. 2008, 196, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Dobson, G.P.; Letson, H.L.; Sharma, R.; Sheppard, F.R.; Cap, A.P. Mechanisms of early trauma-induced coagulopathy: The plot thickens or not? J. Trauma Acute Care Surg. 2015, 79, 301–309. [Google Scholar] [CrossRef]
- Simmons, J.W.; Powell, M.F. Acute traumatic coagulopathy: Pathophysiology and resuscitation. Br. J. Anaesth. 2016, 117 (Suppl. S3), iii31–iii43. [Google Scholar] [CrossRef]
- Davenport, R.; Manson, J.; De’Ath, H.; Platton, S.; Coates, A.; Allard, S.; Hart, D.; Pearse, R.; Pasi, K.J.; MacCallum, P.; et al. Functional definition and characterization of acute traumatic coagulopathy. Crit. Care Med. 2011, 39, 2652–2658. [Google Scholar] [CrossRef]
- Pfeifer, R.; Tarkin, I.S.; Rocos, B.; Pape, H. Patterns of mortality and causes of death in polytrauma patients—Has anything changed? Injury 2009, 40, 907–911. [Google Scholar] [CrossRef]
- Matijevic, N.; Wang, Y.W.; Wade, C.E.; Holcomb, J.B.; Cotton, B.A.; Schreiber, M.A.; Muskat, P.; Fox, E.E.; Del Junco, D.J.; Cardenas, J.C.; et al. Cellular microparticle and thrombogram phenotypes in the Prospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study: Correlation with coagulopathy. Thromb. Res. 2014, 134, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Tabaie, S.; Ivascu, N. Viscoelastic testing inside and beyond the operating room. J. Thorac. Dis. 2017, 9 (Suppl. S4), S299–S308. [Google Scholar] [CrossRef]
- Cohen, M.J.; Kutcher, M.; Redick, B.; Nelson, M.; Call, M.; Knudson, M.M.; Schreiber, M.A.; Bulger, E.M.; Muskat, P.; Alarcon, L.H.; et al. Clinical and mechanistic drivers of acute traumatic coagulopathy. J. Trauma Acute Care Surg. 2013, 75 (Suppl. S1), S40–S47. [Google Scholar] [CrossRef]
- Maegele, M.; Schöchl, H.; Cohen, M.J. An update on the coagulopathy of trauma. Shock 2014, 41 (Suppl. S1), 21–25. [Google Scholar] [CrossRef]
- Meizoso, J.P.; Barrett, C.D.; Moore, E.E.; Moore, H.B. Advances in the Management of Coagulopathy in Trauma: The Role of Viscoelastic Hemostatic Assays across All Phases of Trauma Care. Semin. Thromb. Hemost. 2022, 48, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Cohen, J.C.; Davenport, R.A. Acute coagulopathy of trauma: Mechanism, identification and effect. Curr. Opin. Crit. Care 2007, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Martini, W.Z.; Dubick, M.A.; Pusateri, A.E.; Park, M.S.; Ryan, K.L.; Holcomb, J.B. Does bicarbonate correct coagulation function impaired by acidosis in swine? J. Trauma 2006, 61, 99–106. [Google Scholar] [CrossRef]
- Martini, W.Z.; Dubick, M.A.; Wade, C.E.; Holcomb, J.B. Evaluation of tris-hydroxymethylaminomethane on reversing coagulation abnormalities caused by acidosis in pigs. Crit. Care Med. 2007, 35, 1568. [Google Scholar] [CrossRef]
- Tsuei, B.J.; Kearney, P.A. Hypothermia in the trauma patient. Injury 2004, 35, 7–15. [Google Scholar] [CrossRef]
- Shafi, S.; Elliott, A.C.; Gentilello, L. Is hypothermia simply a marker of shock and injury severity or an independent risk factor for mortality in trauma patients? Analysis of a large national trauma registry. J. Trauma 2005, 59, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, M.A. Coagulopathy in the trauma patient. Curr. Opin. Crit. Care 2005, 11, 590–597. [Google Scholar] [CrossRef]
- Schöchl, H.; Schmitt, F.C.F.; Maegele, M. Pathophysiology of Trauma-Induced Coagulopathy. Hamostaseologie 2024, 44, 31–39. [Google Scholar] [CrossRef]
- Thielen, O.; Mitra, S.; Debot, M.; Schaid, T.; Hallas, W.; Gallagher, L.T.; Erickson, C.; Cralley, A.; Stafford, P.; Silliman, C.; et al. Mitigation of trauma-induced endotheliopathy by activated protein C: A potential therapeutic for postinjury thromboinflammation. J. Trauma Acute Care Surg. 2024, 96, 116–122. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Sebghati, J.; Thelin, E.P. Hemostatic disturbances in traumatic brain injury: From mechanism to management. Acta Neurochir. 2025, 167, 146. [Google Scholar] [CrossRef]
- Gando, S.; Tedo, I.; Kubota, M. Posttrauma coagulation and fibrinolysis. Crit. Care Med. 1992, 20, 594. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Cole, E.; Dünser, M.; Bouzat, P.; Gauss, T. Acute traumatic coagulopathy: What you should know, what is debated and what should come next. Anaesth. Crit Care Pain Med. 2025, 101543. [Google Scholar] [CrossRef]
- Shaz, B.H.; Winkler, A.M.; James, A.B.; Hillyer, C.D.; MacLeod, J.B. Pathophysiology of early trauma-induced coagulopathy: Emerging evidence for hemodilution and coagulation factor depletion. J. Trauma 2011, 70, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Sørensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit. Care 2011, 15, R272. [Google Scholar] [CrossRef]
- Costantini, T.W.; Kornblith, L.Z.; Pritts, T.; Coimbra, R. The intersection of coagulation activation and inflammation after injury: What you need to know. J. Trauma Acute Care Surg. 2024, 96, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, J.C.; Matijevic, N.; Baer, L.A.; Holcomb, J.B.; Cotton, B.A.; Wade, C.E. Elevated tissue plasminogen activator and reduced plasminogen activator inhibitor promote hyperfibrinolysis in trauma patients. Shock 2014, 41, 514–521. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Lawson, P.J.; Gonzalez, E.; Fragoso, M.; Morton, A.P.; Gamboni, F.; Chapman, M.P.; Sauaia, A.; Banerjee, A.; et al. Fibrinolysis shutdown phenotype masks changes in rodent coagulation in tissue injury versus hemorrhagic shock. Surgery 2015, 158, 386–392. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Gonzalez, E.; Chapman, M.P.; Chin, T.L.; Silliman, C.C.; Banerjee, A.; Sauaia, A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: The spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J. Trauma Acute Care Surg. 2014, 77, 811–817. [Google Scholar] [CrossRef]
- Naß, J.; Terglane, J.; Gerke, V. Weibel Palade Bodies: Unique Secretory Organelles of Endothelial Cells that Control Blood Vessel Homeostasis. Front. Cell Dev. Biol. 2021, 9, 813995. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huber, D.; Cramer, E.M.; Kaufmann, J.E.; Meda, P.; Massé, J.M.; Kruithof, E.K.; Vischer, U.M. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood 2002, 99, 3637–3645. [Google Scholar] [CrossRef]
- Moore, E.E.; Moore, H.B.; Gonzalez, E.; Sauaia, A.; Banerjee, A.; Silliman, C.C. Rationale for the selective administration of tranexamic acid tinhibit fibrinolysis in the severely injured patient. Transfusion 2016, 56 (Suppl S2), S110–S114. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.H.; Rappold, J.F.; Badloe, J.F.; Berséus, O.; Blackbourne, L.; Brohi, K.H.; Butler, F.K.; Cap, A.P.; Cohen, M.J.; Davenport, R.; et al. Trauma hemostasis and oxygenation research position paper on remote damage control resuscitation: Definitions, current practice, and knowledge gaps. Shock 2014, 41 (Suppl. S1), 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Call, M.; Nelson, M.; Calfee, C.S.; Esmon, C.T.; Brohi, K.; Pittet, J.F. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012, 255, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Ostrowski, S.R. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann Surg. 2017, 265, 597–603. [Google Scholar] [CrossRef]
- Haywood-Watson, R.J.; Holcomb, J.B.; Gonzalez, E.A.; Peng, Z.; Pati, S.; Park, P.W.; Wang, W.; Zaske, A.M.; Menge, T.; Kozar, R.A. Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS ONE 2011, 6, e23530. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011, 254, 194–200. [Google Scholar] [CrossRef]
- Zeineddin, A.; Wu, F.; Chao, W.; Zou, L.; Vesselinov, R.; Chipman, A.M.; Dong, J.F.; Huang, H.; Pati, S.; Kozar, R.A. Biomarkers of endothelial cell dysfunction persist beyond resuscitation in patients with hemorrhagic shock. J. Trauma Acute Care Surg. 2022, 93, 572–578. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, E.G.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.M.; Abou-Khalil, E.; Gaines, B.A.; Leeper, C.M. Endotheliopathy of trauma in children: The association of syndecan-1 with injury and poor outcomes. J. Trauma Acute Care Surg. 2024, 96, 566–572. [Google Scholar] [CrossRef]
- Suzuki, K. Thrombomodulin: A key regulator of intravascular blood coagulation, fibrinolysis, and inflammation, and a treatment for disseminated intravascular coagulation. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2025, 101, 75–97. [Google Scholar] [CrossRef]
- Matsumoto, H.; Annen, S.; Mukai, N.; Ohshita, M.; Murata, S.; Harima, Y.; Ogawa, S.; Okita, M.; Nakabayashi, Y.; Kikuchi, S.; et al. Circulating Syndecan-1 Levels Are Associated with Chronological Coagulofibrinolytic Responses and the Development of Disseminated Intravascular Coagulation (DIC) after Trauma: A Retrospective Observational Study. J. Clin. Med. 2023, 12, 4386. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Yin, S.; Sun, Z.; Pan, Y. Time course of soluble P-selectin and von Willebrand factor levels in trauma patients: A prospective observational study. Scand. J. Trauma Resusc. Emerg. Med. 2013, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, T.A.; Goswami, J.; Moon Tasson, L.; Tischer, A.; Bailey, K.R.; Spears, G.M.; Dong, J.F.; Auton, M.; Kozar, R.; Park, M.S. Quantification of von Willebrand factor and ADAMTS-13 after traumatic injury: A pilot study. Trauma Surg. Acute Care Open 2021, 6, e000703. [Google Scholar] [CrossRef]
- Zeineddin, A.; Dong, J.F.; Wu, F.; Terse, P.; Kozar, R.A. Role of Von Willebrand Factor after Injury: It May Do More Than We Think. Shock 2021, 55, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.R.; Plautz, W.E.; Ragni, M.V.; Alexander, W.; Haldeman, S.; Sperry, J.L.; Guyette, F.X.; Zuckerbraun, B.S.; Rollins-Raval, M.A.; Raval, J.S.; et al. A TACTIC Publication. Traumatic injury results in prolonged circulation of ultralarge von Willebrand factor and a reduction in ADAMTS13 activity. Transfusion 2020, 60, 1308–1318. [Google Scholar] [CrossRef]
- Schaid, T.R., Jr.; Mitra, S.; Stafford, P.; DeBot, M.; Thielen, O.; Hallas, W.; Cralley, A.; Gallagher, L.; Jeffrey, D.; Hansen, K.C.; et al. Endothelial Cell Calcium Influx Mediates Trauma-induced Endothelial Permeability. Ann. Surg. 2025, 281, 671–681. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Huang, Y.; Yao, X. TRP channels in endothelial function and dysfunction. Biochim. Biophys. Acta 2007, 1772, 907–914. [Google Scholar] [CrossRef]
- DeBot, M.; Mitra, S.; Lutz, P.; Schaid, T.R., Jr.; Stafford, P.; Hadley, J.B.; Hom, P.; Sauaia, A.; Silliman, C.C.; Moore, E.E.; et al. SHOCK INDUCES ENDOTHELIAL PERMEABILITY AFTER TRAUMA THROUGH INCREASED ACTIVATION OF RHOA GTPASE. Shock 2022, 58, 542–548. [Google Scholar] [CrossRef]
- Ho, J.W.; Dawood, Z.S.; Taylor, M.E.; Liggett, M.R.; Jin, G.; Jaishankar, D.; Nadig, S.N.; Bharat, A.; Alam, H.B. The neuroendothelial axis in traumatic brain injury: Mechanisms of multiorgan dysfunction, novel therapies and future directions. Shock 2024, 61, 346–359. [Google Scholar] [CrossRef]
- Yang, Z.; Le, T.D.; Simovic, M.O.; Liu, B.; Fraker, T.L.; Cancio, T.S.; Cap, A.P.; Wade, C.E.; DalleLucca, J.J.; Li, Y. Traumatized triad of complementopathy, endotheliopathy, and coagulopathy—Impact on clinical outcomes in severe polytrauma patients. Front. Immunol. 2022, 13, 991048. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Potter, D.R.; Miyazawa, B.Y.; Gibb, S.L.; Deng, X.; Togaratti, P.P.; Croze, R.H.; Srivastava, A.K.; Trivedi, A.; Matthay, M.; Holcomb, J.B.; et al. Mesenchymal stem cell-derived extracellular vesicles attenuate pulmonary vascular permeability and lung injury induced by hemorrhagic shock and trauma. J. Trauma Acute Care Surg. 2018, 84, 245–256. [Google Scholar] [CrossRef]
- Li, F.; Li, L.; Peng, R.; Liu, C.; Liu, X.; Liu, Y.; Wang, C.; Xu, J.; Zhang, Q.; Yang, G.; et al. Brain-derived extracellular vesicles mediate systemic coagulopathy and inflammation after traumatic brain injury. Int. Immunopharmacol. 2024, 130, 111674. [Google Scholar] [CrossRef]
- Li, L.; Li, F.; Bai, X.; Jia, H.; Wang, C.; Li, P.; Zhang, Q.; Guan, S.; Peng, R.; Zhang, S.; et al. Circulating extracellular vesicles from patients with traumatic brain injury induce cerebrovascular endothelial dysfunction. Pharmacol. Res. 2023, 192, 106791. [Google Scholar] [CrossRef]
- Ng, W.; Jerath, A.; Wąsowicz, M. Tranexamic acid: A clinical review. Anaesthesiol. Intensive Ther. 2015, 47, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Relke, N.; Chornenki, N.L.J.; Sholzberg, M. Tranexamic acid evidence and controversies: An illustrated review. Res. Pract. Thromb. Haemost. 2021, 5, e12546. [Google Scholar] [CrossRef]
- Colomina, M.J.; Contreras, L.; Guilabert, P.; Koo, M.; MNdez, E.; Sabate, A. Clinical use of tranexamic acid: Evidences and controversies. Braz. J Anesthesiol. 2022, 72, 795–812. [Google Scholar] [CrossRef] [PubMed]
- CRASH-2 Collaborators; Roberts, I.; Shakur, H. The importance of early treatment with tranexamic acid in bleeding trauma patients: An exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011, 377, 1096–1101, 1101.e1–1101.e2. [Google Scholar]
- Morrison, J.J.; Dubose, J.J.; Rasmussen, T.E. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) Study. Arch. Surg. 2012, 147, 113–119. [Google Scholar] [CrossRef]
- Spahn, D.R.; Bouillon, B.; Cerny, V. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar]
- Del Junco, D.J.; Holcomb, J.B.; Fox, E.E. Resuscitate early with plasma and platelets or balance blood products gradually: Findings from the PROMMTT study. J. Trauma Acute Care Surg. 2013, 75, S24. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, J.B.; Tilley, B.C.; Baraniuk, S. Transfusion of plasma, platelets, and red blood cells in a 1, 1, 1 vs. a 1, 1, 2 ratio and mortality in patients with severe trauma: The PROPPR randomized clinical trial. JAMA 2015, 313, 471. [Google Scholar] [CrossRef]
- Gonzalez, E.; Moore, E.E.; Moore, H.B. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: A pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann. Surg. 2016, 263, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Stettler, G.R.; Sumislawski, J.J.; Moore, E.E.; Nunns, G.R.; Kornblith, L.Z.; Conroy, A.S.; Callcut, R.A.; Silliman, C.C.; Banerjee, A.; Cohen, M.J.; et al. Citrated kaolin thrombelastography (TEG) thresholds for goal-directed therapy in injured patients receiving massive transfusion. J. Trauma Acute Care Surg. 2018, 85, 734–740. [Google Scholar] [CrossRef]
- Brill, J.B.; Brenner, M.; Duchesne, J.; Roberts, D.; Ferrada, P.; Horer, T.; Kauvar, D.; Khan, M.; Kirkpatrick, A.; Ordonez, C.; et al. The Role of TEG and ROTEM in Damage Control Resuscitation. Shock 2021, 56, 52–61. [Google Scholar] [CrossRef]
- Stevens, J.; Pickett, K.; Moore, H.; Reppucci, M.L.; Phillips, R.; Moulton, S.; Bensard, D. Thrombelastography and transfusion patterns in severely injured pediatric trauma patients with blunt solid organ injuries. J. Trauma Acute Care Surg. 2022, 92, 152–158. [Google Scholar] [CrossRef]
- Dudek, C.J.; Little, I.; Wiser, K.; Ibrahim, J.; Ramirez, J.; Papa, L. Thromboelastography Use in the Acute Young Trauma Patient: Early Experience of Two Level One Trauma Centers. Injury 2021, 52, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hagemo, J.S.; Christiaans, S.C.; Stanworth, S.J.; Brohi, K.; Johansson, P.I.; Goslings, J.C.; Naess, P.A.; Gaarder, C. Detection of acute traumatic coagulopathy and massive transfusion requirements by means of rotational thromboelastometry: An international prospective validation study. Crit. Care 2015, 23, 97. [Google Scholar] [CrossRef]
- Hutspardol, S.; Borja, T.; Kroeker, J.; Wang, X.Q.; Mi, J.; Zamar, D.; Chan, G.; Smith, T.; Hawes, H.; Shih, A.W. Comparison of conventional coagulation tests and ROTEM in identifying trauma-induced coagulopathy for massive haemorrhage protocol activation. Transfus. Med. 2025. online ahead of print. [Google Scholar] [CrossRef]
- Nascimento, B.; Callum, J.; Tien, H. Fibrinogen in the initial resuscitation of severe trauma (FiiRST): A randomized feasibility trial. Br. J. Anaesth. 2016, 117, 775–782. [Google Scholar] [CrossRef]
- Rizoli, S.B.; Boffard, K.D.; Riou, B. Recombinant activated factor VII as an adjunctive therapy for bleeding control in severe trauma patients with coagulopathy: Subgroup analysis from two randomized trials. Crit. Care 2006, 10, R178. [Google Scholar] [CrossRef] [PubMed]
- Dickneite, G.; Dörr, B.; Kaspereit, F.; Tanaka, K.A. Prothrombin complex concentrate versus recombinant factor VIIa for reversal of hemodilutional coagulopathy in a porcine trauma model. J. Trauma 2010, 68, 1151. [Google Scholar] [CrossRef]
- Joseph, B.; Hadjizacharia, P.; Aziz, H.; Kulvatunyou, N.; Tang, A.; Pandit, V.; Wynne, J.; O’Keeffe, T.; Friese, R.S.; Rhee, P. Prothrombin complex concentrate: An effective therapy in reversing the coagulopathy of traumatic brain injury. J. Trauma 2013, 74, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.; Hirsh, J.; Poller, L. The pharmacology and management of the vitamin K antagonists: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004, 126, 204S–233S. [Google Scholar] [CrossRef]
- Spinella, P.C.; Holcomb, J.B. Resuscitation and transfusion principles for traumatic hemorrhagic shock. Blood Rev. 2009, 23, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.J.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef]
- Salmon, A.H.J.; Neal, C.R.; Sage, L.M.; Glass, C.A.; Harper, S.J.; Bates, D.O. Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc. Res. 2009, 83, 24–33. [Google Scholar] [CrossRef]
- Chappell, D.; Dörfler, N.; Jacob, M.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Glicocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 2010, 34, 133–139. [Google Scholar] [CrossRef]
- Del Pilar Huby Vidaurre, M.; Mokhtari, A.K.; Osborn, B.K.; Cotton, B.A.; Wang, Y.W.; Xu, Y.; Arnold, K.; Liu, J.; Richter, J.R.; Cardenas, J.C. The interaction between antithrombin and endothelial heparan sulfate mitigates pulmonary thromboinflammation after trauma and hemorrhagic shock. Shock 2025, 63, 638–647. [Google Scholar] [CrossRef]
- Lipowsky, H.H.; Lescanic, A. Inhibition of inflammation induced shedding of the endothelial glycocalyx with low molecular weight heparin. Microvasc. Res. 2017, 112, 72–78. [Google Scholar] [CrossRef]
- Xu, X.; Kozar, R.; Zhang, J.; Dong, J.F. Diverse activities of von Willebrand factor in traumatic brain injury and associated coagulopathy. J. Thromb. Haemost. 2020, 18, 3154–3162. [Google Scholar] [CrossRef] [PubMed]
- Kleinveld, D.J.B.; Simons, D.D.G.; Dekimpe, C.; Deconinck, S.J.; Sloos, P.H.; Maas, M.A.W.; Kers, J.; Muia, J.; Brohi, K.; Voorberg, J.; et al. Plasma and rhADAMTS13 reduce trauma-induced organ failure by restoring the ADAMTS13-VWF axis. Blood Adv. 2021, 5, 3478–3491. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Vendramin, C.; Singh, D.; Brown, M.M.; von Scully, M. Willebrand factor/ADAMTS13 ratio at presentation of acute ischemic brain injury is pre- dictive of outcome. Blood Adv. 2020, 4, 398–407. [Google Scholar] [CrossRef]
- Plautz, W.E.; Raval, J.S.; Dyer, M.R.; Rollins-Raval, M.A.; Zuckerbraun, B.S.; Neal, M.D. ADAMTS13: Origins, applications, and prospects. Transfusion 2018, 58, 2453–2462. [Google Scholar] [CrossRef]
- Russell, R.T.; McDaniel, J.K.; Cao, W.; Shroyer, M.; Wagener, B.M.; Zheng, X.L.; Pittel, J.F. Low plasma ADAMTS13 activity is associated with coagulopathy, en- dothelial cell damage and mortality after severe paediatric trauma. Thromb. Haemost. 2018, 118, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Jiang, S.; Guo, J.; Xu, N.; Wang, Q.; Zhang, G.; Pittet, J.F. ADAMTS13 protects mice against renal ischemia-reperfusion injury by reducing inflammation and improving endothelial function. Am. J. Physiol. Renal. Physiol. 2019, 316, F134–F145. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, W.; Zhou, Y.; Hilton, T.; Zhao, Z.; Liu, W.; Wang, M.; Yeon, J.; Houck, K.; Thiagarajan, P.; et al. von Willebrand factor enhances microvesicle-induced vascular leakage and coagulopathy in mice with traumatic brain injury. Blood 2018, 132, 1075–1084. [Google Scholar] [CrossRef]
- Wang, J.; Xie, X.; Wu, Y.; Zhou, Y.; Li, Q.; Li, Y.; Xu, X.; Wang, M.; Murdiyarso, L.; Houck, K.; et al. Brain-Derived Extracellular Vesicles Induce Vasoconstriction and Reduce Cerebral Blood Flow in Mice. J Neurotrauma 2022, 39, 879–890. [Google Scholar] [CrossRef]
- Zeineddin, A.; Wu, F.; Dong, J.F.; Huang, H.; Zou, L.; Chao, W.; Dorman, B.; Kozar, R.A. Trauma-derived extracellular vesicles are sufficient to induce endothelial dysfunction and coagulopathy. Shock 2022, 58, 38–44. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, W.; Zhao, Z.; Hilton, T.; Wang, M.; Yeon, J.; Liu, W.; Zhang, F.; Shi, F.D.; Wu, X.; et al. Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of severe TBI mice. Blood 2018, 131, 563–572. [Google Scholar] [CrossRef]
- Matthay, M.A.; Pati, S.; Lee, J.W. Concise review: Mesenchymal stem (stromal) cells: Biology and preclinical evidence for therapeutic potential for organ dysfunction following trauma or sepsis. Stem Cells 2017, 35, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.; Trivedi, A.; Pathipati, P.; Miyazawa, B.Y.; Vivona, L.R.; Togarrati, P.P.; Khakoo, M.; Tanner, H.; Norris, P.; Pati, S. Mesenchymal stem cell extracellular vesicles mitigate vascular permeability and injury in the small intestine and lung in a mouse model of hemorrhagic shock and trauma. J. Trauma Acute Care Surg. 2022, 92, 489–498. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tranexamic acid | Inhibits fibrinolysis through competitive and non-competitive plasmin inhibition |
| Goal-Direct transfusion | Balanced transfusion protocol with equal parts plasma, RBCs, and platelets (1:1:1 ratio) can enhance survival |
| Fibrinogen | Guidelines recommend fibrinogen replacement when levels drop below 1.5 g/L during significant bleeding |
| Calcium chloride | Restoration of normal calcium levels since calcium have an essential role in the formation and stabilization of fibrin polymerization sites and on platelet function |
| Etanercept angiopoietin-1 | Modulation of EGL shedding |
| Doxycylcyne (MMP inhibitor) | Reduction of glycocalyx shedding |
| Dekaparin (synthetic 3.0 heparan sulfate) | Modulation of coagulation factors activation, anti-inflammatory effects on endothelial surface |
| Recombinant-human ADAMTS 13 | Decreased coagulopathy and endothelial permeability and induced organ damage |
| Lactadherin | Facilitate removal of extracellular vesicles |
| Mesenchymal stem cells | Promotion of angiogenesis and decrease in endothelial permeability |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capponi, A.; Rostagno, C. Trauma-Induced Coagulopathy: A Review of Specific Molecular Mechanisms. Diagnostics 2025, 15, 1435. https://doi.org/10.3390/diagnostics15111435
Capponi A, Rostagno C. Trauma-Induced Coagulopathy: A Review of Specific Molecular Mechanisms. Diagnostics. 2025; 15(11):1435. https://doi.org/10.3390/diagnostics15111435
Chicago/Turabian StyleCapponi, Andrea, and Carlo Rostagno. 2025. "Trauma-Induced Coagulopathy: A Review of Specific Molecular Mechanisms" Diagnostics 15, no. 11: 1435. https://doi.org/10.3390/diagnostics15111435
APA StyleCapponi, A., & Rostagno, C. (2025). Trauma-Induced Coagulopathy: A Review of Specific Molecular Mechanisms. Diagnostics, 15(11), 1435. https://doi.org/10.3390/diagnostics15111435