



Meta-Analysis of Wild Relatives and Domesticated Species of Rice, Tomato, and Soybean Using Publicly Available Transcriptome Data



Abstract
1. Introduction
2. Materials and Methods
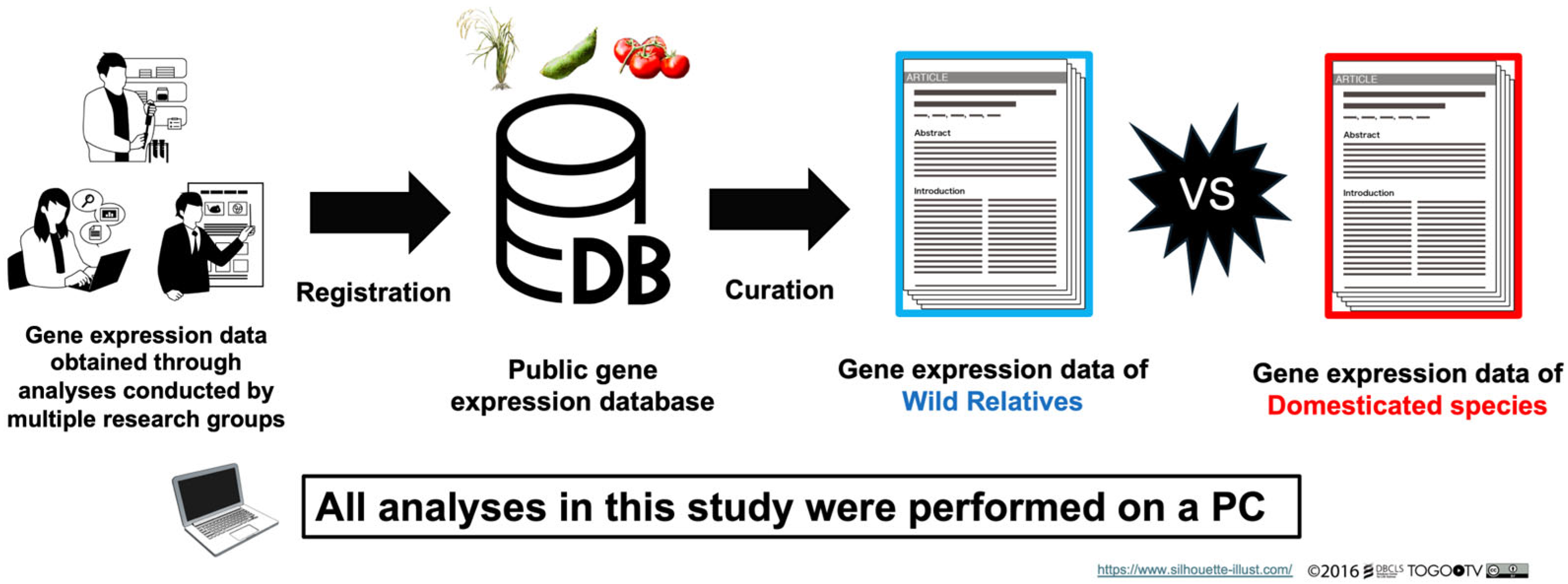
2.1. Curation of Public Gene Expression Data
2.2. Gene Expression Quantification
2.3. Calculation of the DW Ratio
2.4. Classification of Differentially Expressed Genes (DEGs) Based on the DW Ratio
2.5. Calculation of the DW Score
2.6. Gene Set Enrichment Analysis
2.7. Commonly Upregulated Genes in Wild and Domesticated Species
3. Results
3.1. Overview of the Study
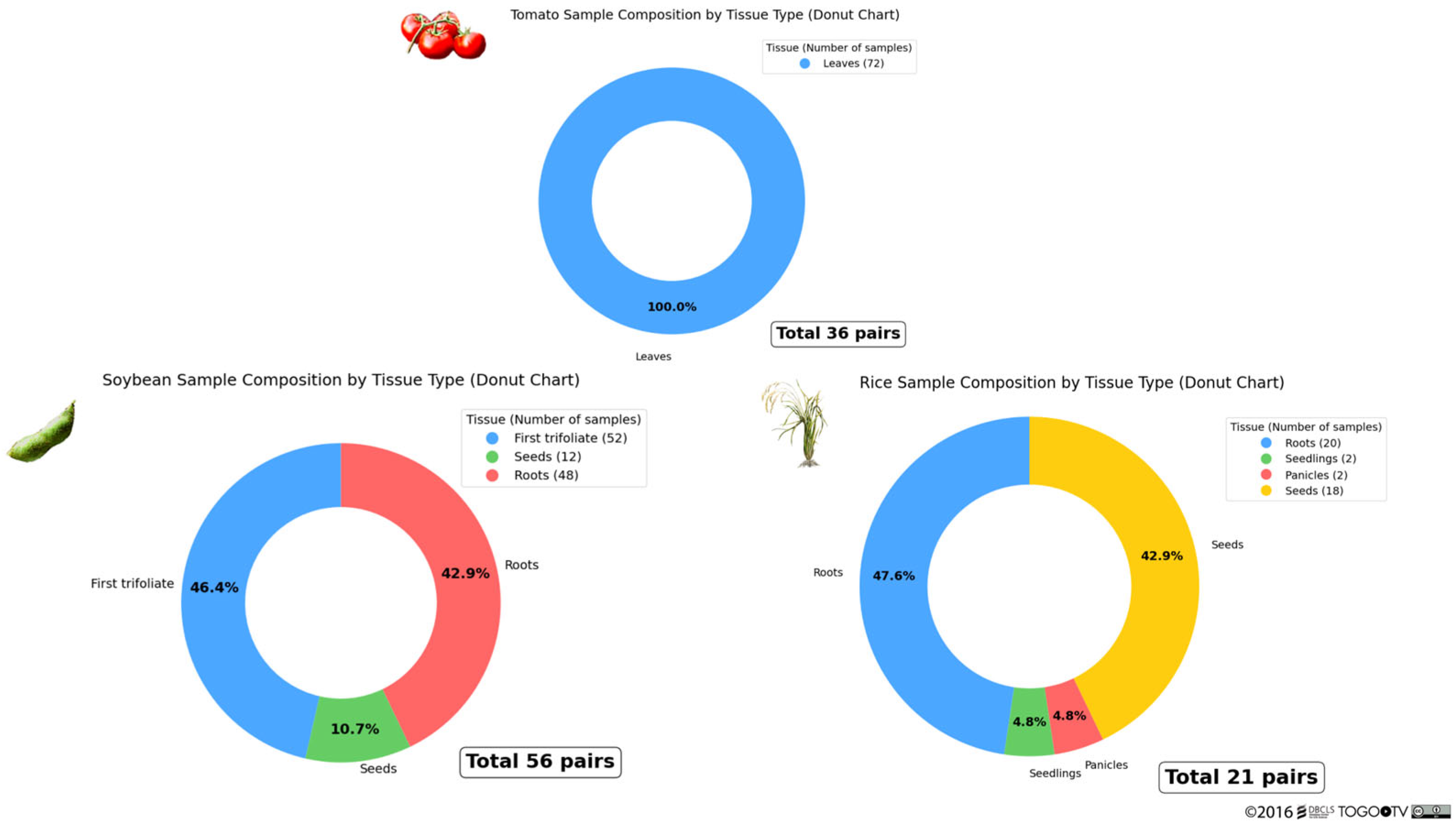
3.2. Curation of RNA-Seq Data from Public Databases and the Literature
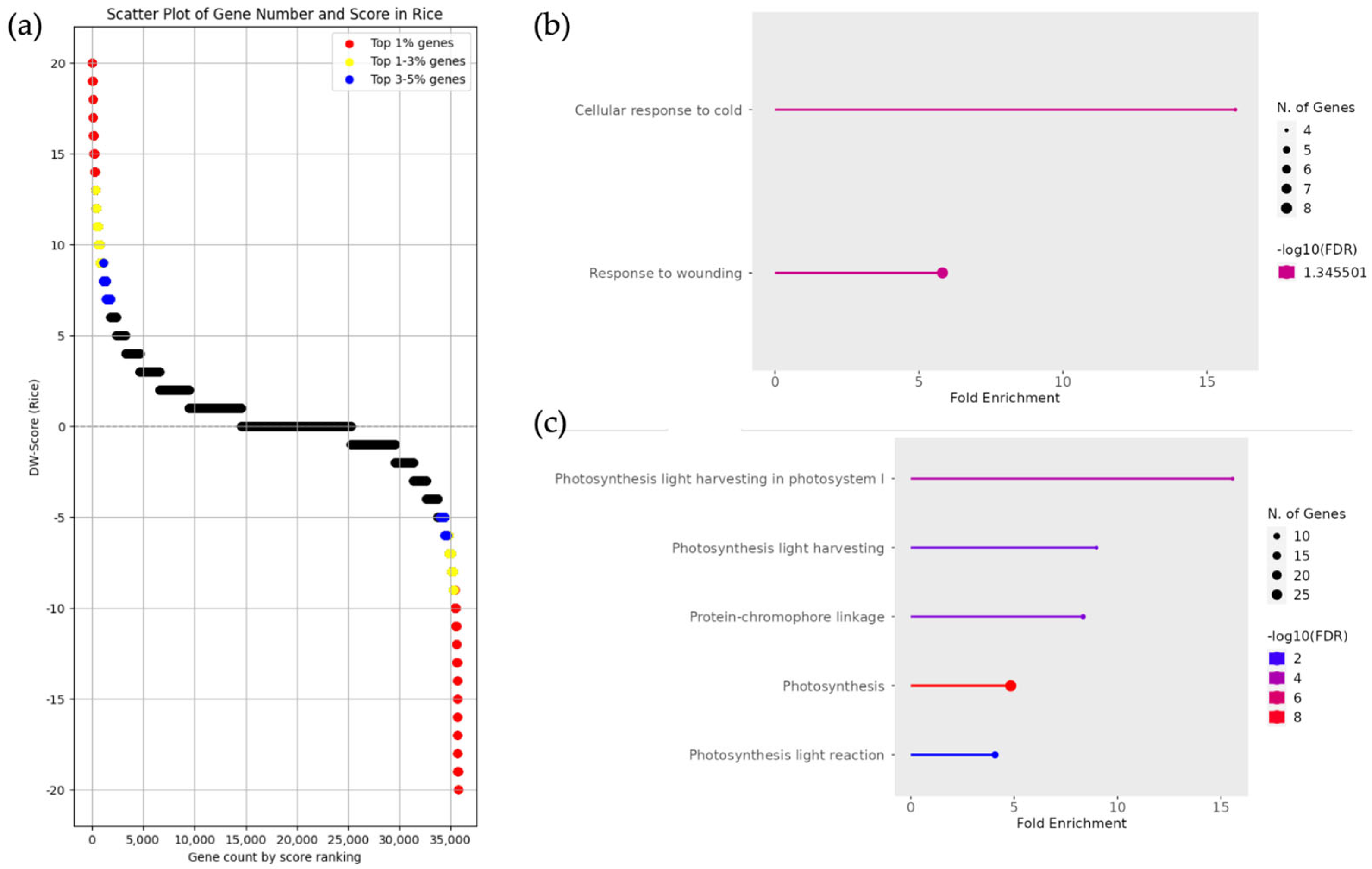
3.3. Classification of DEGs and Enrichment Analysis in Oryza rufipogon and Oryza sativa japonica
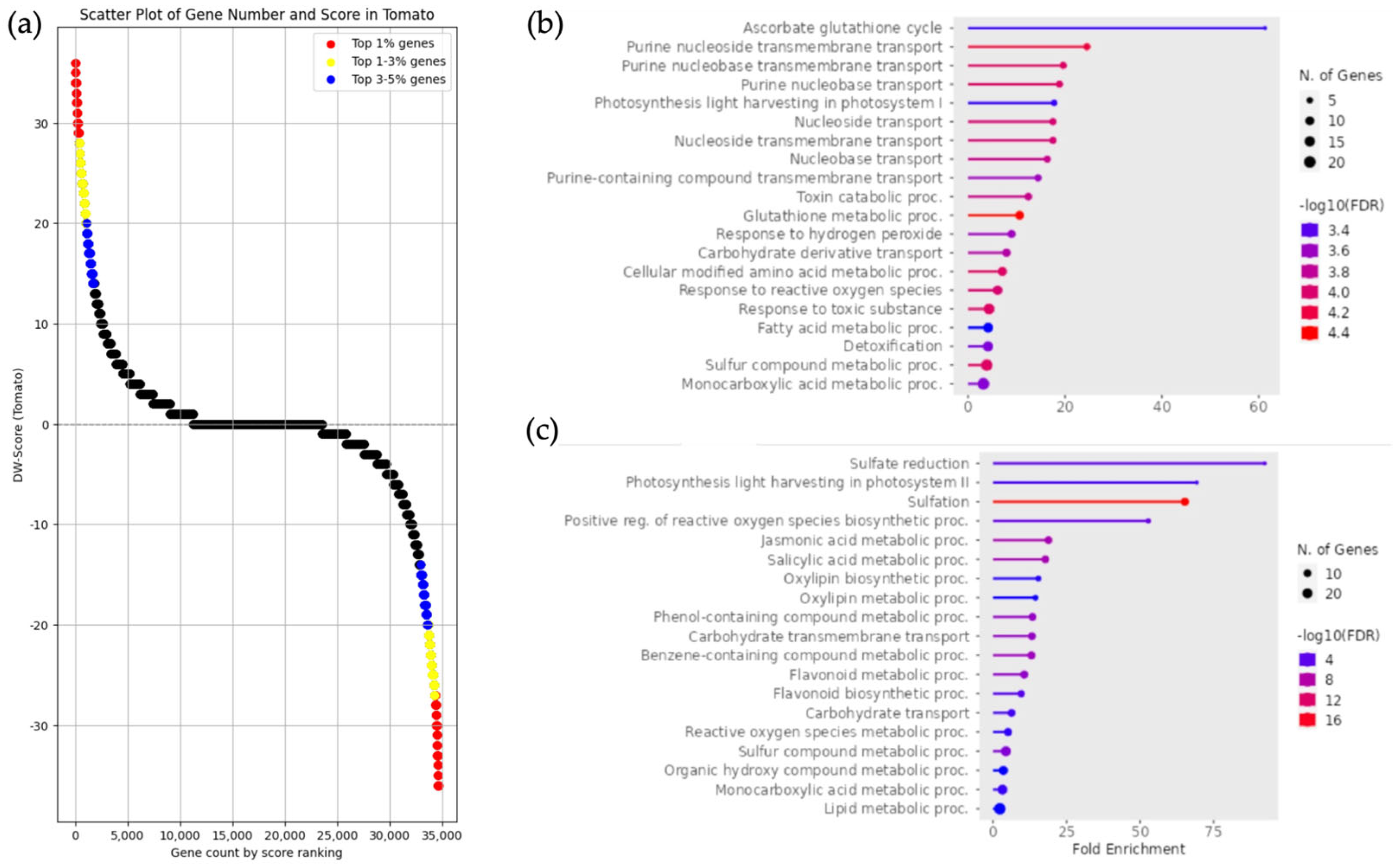
3.4. Classification of DEGs and Enrichment Analysis of Solanum pennellii, Solanum arcanum, and Solanum lycopersicum
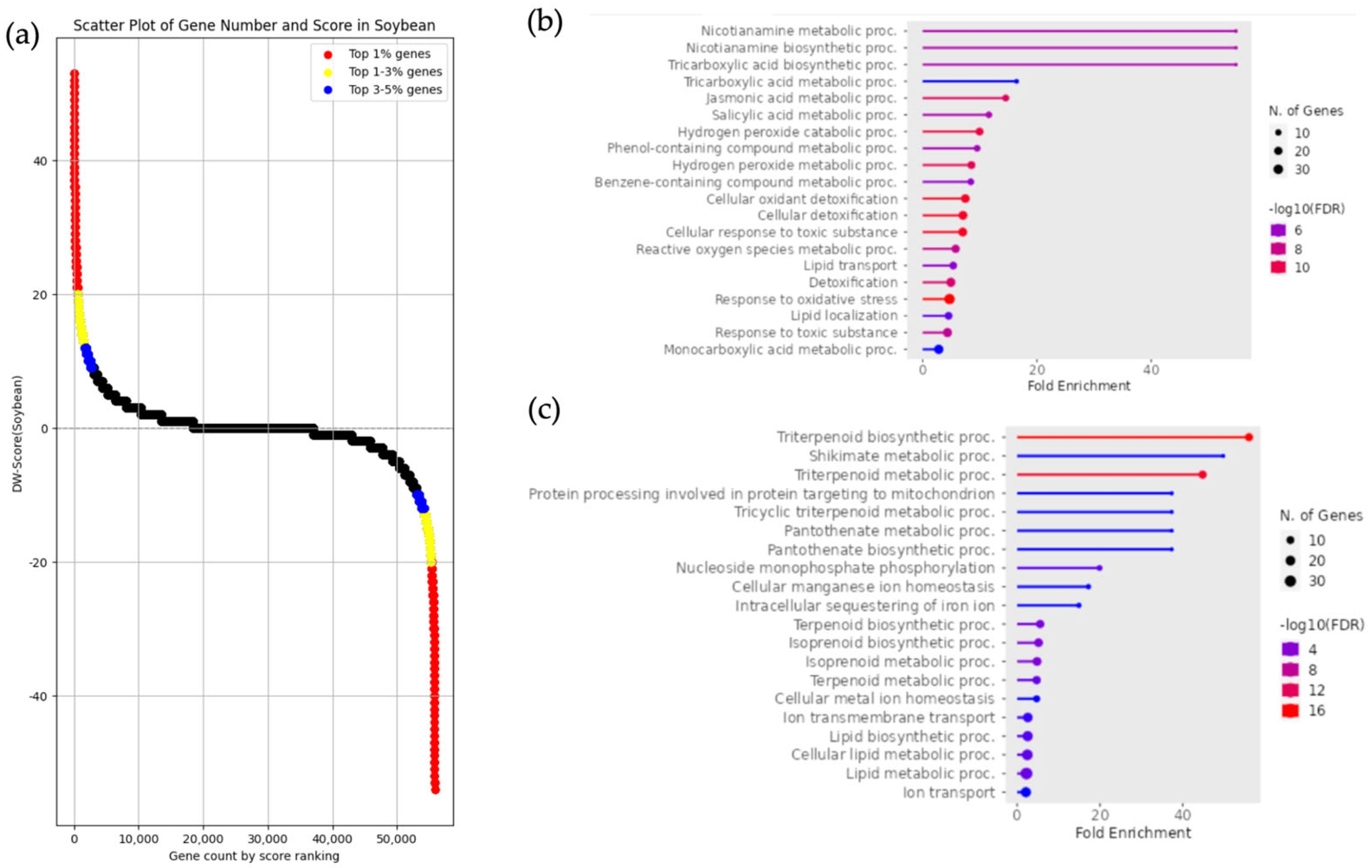
3.5. Classification of DEGs and Enrichment Analysis in Glycine soja and Glycine max
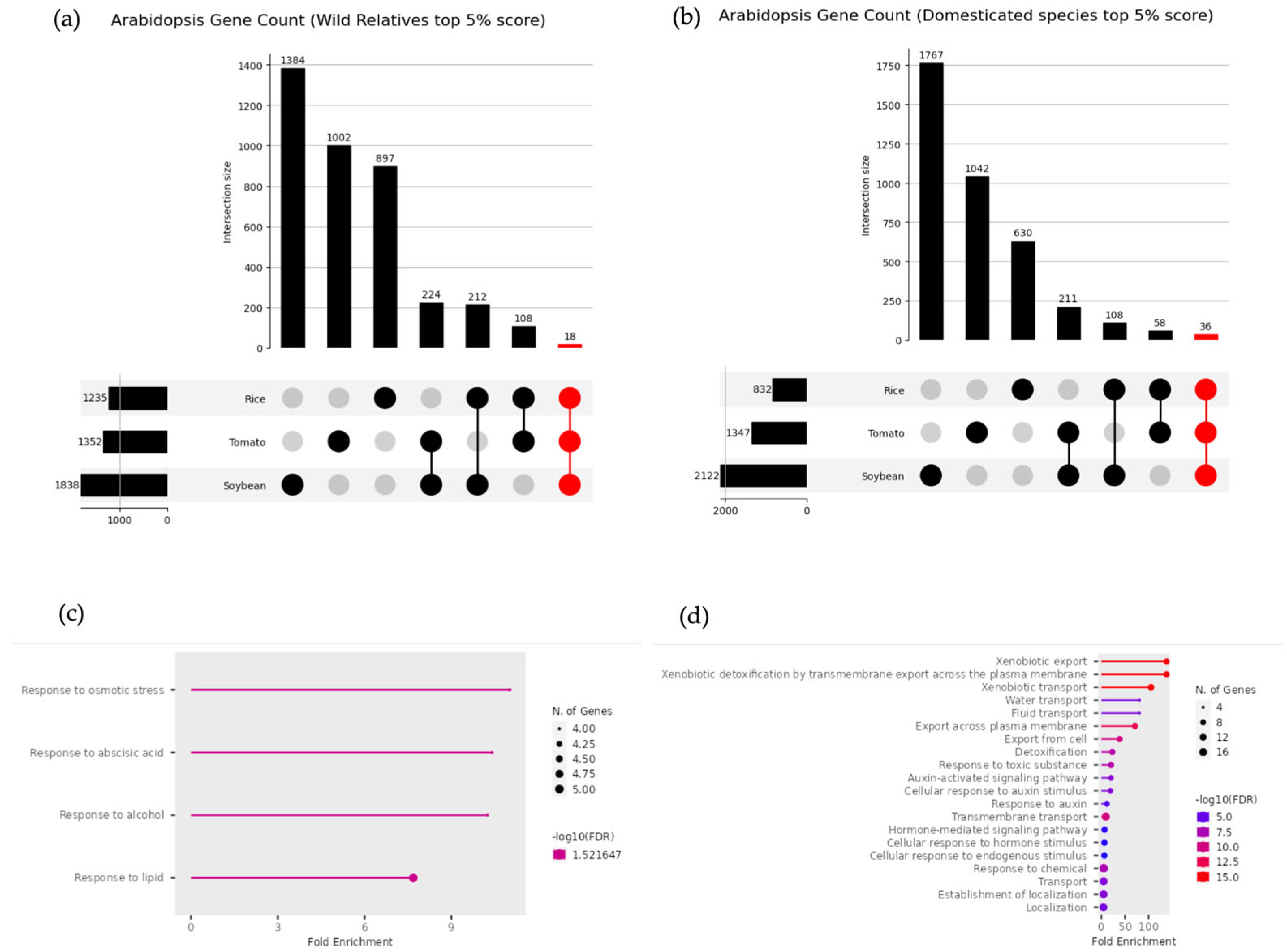
3.6. Common DEGs in Wild and Domesticated Species
4. Discussion
4.1. Genes Commonly Upregulated Across Wild Relatives
4.2. Genes Commonly Upregulated Across Domesticated Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA-seq | RNA sequencing |
| NCBI GEO | National Center for Biotechnology Information Gene Expression Omnibus |
| TPM | Transcripts per million |
| DW | Domesticated and wild |
| DEG | Differentially expressed gene |
| GO | Gene Ontology |
| ABA | Abscisic acid |
| MATE | Multidrug and toxic compound extrusion |
References
- Challinor, A.J.; Watson, J.; Lobell, D.B.; Howden, S.M.; Smith, D.R.; Chhetri, N. A Meta-Analysis of Crop Yield under Climate Change and Adaptation. Nat. Clim. Change 2014, 4, 287–291. [Google Scholar] [CrossRef]
- Aggarwal, P.; Vyas, S.; Thornton, P.; Campbell, B.M.; Kropff, M. Importance of Considering Technology Growth in Impact Assessments of Climate Change on Agriculture. Glob. Food Secur. 2019, 23, 41–48. [Google Scholar] [CrossRef]
- Lobell, D.B.; Gourdji, S.M. The Influence of Climate Change on Global Crop Productivity. Plant Physiol. 2012, 160, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.; von Braun, J. Climate Change Impacts on Global Food Security. Science 2013, 341, 508–513. [Google Scholar] [CrossRef]
- van Dijk, M.; Morley, T.; Rau, M.L.; Saghai, Y. A Meta-Analysis of Projected Global Food Demand and Population at Risk of Hunger for the Period 2010–2050. Nat. Food 2021, 2, 494–501. [Google Scholar] [CrossRef]
- Kishimoto, K.; Washio, Y.; Yoshiura, Y.; Toyoda, A.; Ueno, T.; Fukuyama, H.; Kato, K.; Kinoshita, M. Production of a Breed of Red Sea Bream Pagrus Major with an Increase of Skeletal Muscle Mass and Reduced Body Length by Genome Editing with CRISPR/Cas9. Aquaculture 2018, 495, 415–427. [Google Scholar] [CrossRef]
- Nonaka, S.; Arai, C.; Takayama, M.; Matsukura, C.; Ezura, H. Efficient Increase of Ɣ-Aminobutyric Acid (GABA) Content in Tomato Fruits by Targeted Mutagenesis. Sci. Rep. 2017, 7, 7057. [Google Scholar] [CrossRef]
- Yu, H.; Lin, T.; Meng, X.; Du, H.; Zhang, J.; Liu, G.; Chen, M.; Jing, Y.; Kou, L.; Li, X.; et al. A Route to de Novo Domestication of Wild Allotetraploid Rice. Cell 2021, 184, 1156–1170.e14. [Google Scholar] [CrossRef]
- Zsögön, A.; Čermák, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De Novo Domestication of Wild Tomato Using Genome Editing. Nat. Biotechnol. 2018, 36, 1211–1216. [Google Scholar] [CrossRef]
- Zsögön, A.; Cermak, T.; Voytas, D.; Peres, L.E.P. Genome Editing as a Tool to Achieve the Crop Ideotype and de Novo Domestication of Wild Relatives: Case Study in Tomato. Plant Sci. 2017, 256, 120–130. [Google Scholar] [CrossRef]
- Krug, A.S.; Drummond, E.B.M.; Van Tassel, D.L.; Warschefsky, E.J. The next Era of Crop Domestication Starts Now. Proc. Natl. Acad. Sci. USA 2023, 120, e2205769120. [Google Scholar] [CrossRef]
- Curtin, S.; Qi, Y.; Peres, L.E.P.; Fernie, A.R.; Zsögön, A. Pathways to de Novo Domestication of Crop Wild Relatives. Plant Physiol. 2022, 188, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, J. Breeding Future Crops to Feed the World through de Novo Domestication. Nat. Commun. 2022, 13, 1171. [Google Scholar] [CrossRef]
- Gasparini, K.; dos Reis Moreira, J.; Peres, L.E.P.; Zsögön, A. De Novo Domestication of Wild Species to Create Crops with Increased Resilience and Nutritional Value. Curr. Opin. Plant Biol. 2021, 60, 102006. [Google Scholar] [CrossRef]
- Razzaq, A.; Saleem, F.; Wani, S.H.; Abdelmohsen, S.A.M.; Alyousef, H.A.; Abdelbacki, A.M.M.; Alkallas, F.H.; Tamam, N.; Elansary, H.O. De-Novo Domestication for Improving Salt Tolerance in Crops. Front. Plant Sci. 2021, 12, 681367. [Google Scholar] [CrossRef]
- Shafiq, M.; Azeem, F.; Waheed, Y.; Pamirsky, I.E.; Feng, X.; Golokhvast, K.S.; Nawaz, M.A. Meta-Analysis of RNA-Seq Data of Soybean under Heat, Water, and Drought Stresses. Plant Biotechnol. Rep. 2025, 19, 205–222. [Google Scholar] [CrossRef]
- Fukuda, Y.; Kawaguchi, K.; Fukushima, A. AtSRGA: A Shiny Application for Retrieving and Visualizing Stress-Responsive Genes in Arabidopsis thaliana. Plant Physiol. 2025, 197, kiaf105. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sragrowth/ (accessed on 1 April 2025).
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets-Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Ncbi/Sra-Tools: SRA Tools. Available online: https://github.com/ncbi/sra-tools (accessed on 5 March 2025).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Tximport. Available online: http://bioconductor.org/packages/tximport/ (accessed on 20 March 2025).
- Yonezawa, S. Yonesora56/HS_rice. (21 December 2023). Jupyter Notebook. [Online]. Available online: https://github.com/yonesora56/HS_rice (accessed on 20 March 2025).
- Ono, Y.; Bono, H. Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes. Biomedicines 2021, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinforma. Oxf. Engl. 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A Hub for Data Retrieval across Taxonomic Space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—from Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; on behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Molina, L.; Mongrand, S.; Chua, N.-H. A Postgermination Developmental Arrest Checkpoint Is Mediated by Abscisic Acid and Requires the ABI5 Transcription Factor in Arabidopsis. Proc. Natl. Acad. Sci. USA 2001, 98, 4782–4787. [Google Scholar] [CrossRef]
- Sah, S.K.; Reddy, K.R.; Li, J. Abscisic Acid and Abiotic Stress Tolerance in Crop Plants. Front. Plant Sci. 2016, 7, 571. [Google Scholar] [CrossRef]
- Finkelstein, R.R.; Lynch, T.J. Abscisic Acid Inhibition of Radicle Emergence But Not Seedling Growth Is Suppressed by Sugars1. Plant Physiol. 2000, 122, 1179–1186. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, J.; Li, Y.; Quan, R.; Wang, J.; Huang, R.; Qin, H. Salt Stress Promotes Abscisic Acid Accumulation to Affect Cell Proliferation and Expansion of Primary Roots in Rice. Int. J. Mol. Sci. 2021, 22, 10892. [Google Scholar] [CrossRef]
- Horie, T.; Hauser, F.; Schroeder, J.I. HKT Transporter-Mediated Salinity Resistance Mechanisms in Arabidopsis and Monocot Crop Plants. Trends Plant Sci. 2009, 14, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Saito, S.; Yamanashi, T.; Kato, M.; Takebayashi, K.; Hamamoto, S.; Tsujii, M.; Takagi, T.; Nagata, N.; Ikeda, H.; et al. The HKT1 Na+ Transporter Protects Plant Fertility by Decreasing Na+ Content in Stamen Filaments. Sci. Adv. 2023, 9, eadg5495. [Google Scholar] [CrossRef]
- Abe, H.; Yamaguchi-Shinozaki, K.; Urao, T.; Iwasaki, T.; Hosokawa, D.; Shinozaki, K. Role of Arabidopsis MYC and MYB Homologs in Drought- and Abscisic Acid-Regulated Gene Expression. Plant Cell 1997, 9, 1859–1868. [Google Scholar] [CrossRef]
- Matus, J.T.; Aquea, F.; Espinoza, C.; Vega, A.; Cavallini, E.; Santo, S.D.; Cañón, P.; Guardia, A.R.-H.; Serrano, J.; Tornielli, G.B.; et al. Inspection of the Grapevine BURP Superfamily Highlights an Expansion of RD22 Genes with Distinctive Expression Features in Berry Development and ABA-Mediated Stress Responses. PLoS ONE 2014, 9, e110372. [Google Scholar] [CrossRef]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) Function as Transcriptional Activators in Abscisic Acid Signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene Expression and Signal Transduction in Water-Stress Response. Plant Physiol. 1997, 115, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Kiyosue, T.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A Nuclear Gene, Erd1, Encoding a Chloroplast-Targeted Clp Protease Regulatory Subunit Homolog Is Not Only Induced by Water Stress but Also Developmentally up-Regulated during Senescence in Arabidopsis thaliana. Plant J. 1997, 12, 851–861. [Google Scholar] [CrossRef]
- Ré, D.A.; Capella, M.; Bonaventure, G.; Chan, R.L. Arabidopsis AtHB7 and AtHB12 Evolved Divergently to Fine Tune Processes Associated with Growth and Responses to Water Stress. BMC Plant Biol. 2014, 14, 150. [Google Scholar] [CrossRef]
- Hamberg, M.; Sanz, A.; Castresana, C. α-Oxidation of Fatty Acids in Higher Plants: Identification of a pathogen-inducible oxygenase (piox) as an α-dioxygenase and biosynthesis of 2-hydroperoxylinolenic acid*. J. Biol. Chem. 1999, 274, 24503–24513. [Google Scholar] [CrossRef]
- De León, I.P.; Sanz, A.; Hamberg, M.; Castresana, C. Involvement of the Arabidopsisα-DOX1 Fatty Acid Dioxygenase in Protection against Oxidative Stress and Cell Death. Plant J. 2002, 29, 61–72. [Google Scholar] [CrossRef]
- Vicente, J.; Cascón, T.; Vicedo, B.; García-Agustín, P.; Hamberg, M.; Castresana, C. Role of 9-Lipoxygenase and α-Dioxygenase Oxylipin Pathways as Modulators of Local and Systemic Defense. Mol. Plant 2012, 5, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Denekamp, M.; Smeekens, S.C. Integration of Wounding and Osmotic Stress Signals Determines the Expression of the AtMYB102 Transcription Factor Gene. Plant Physiol. 2003, 132, 1415–1423. [Google Scholar] [CrossRef]
- De Vos, M.; Denekamp, M.; Dicke, M.; Vuylsteke, M.; Van Loon, L.C.; Smeekens, S.C.; Pieterse, C. The Arabidopsis thaliana Transcription Factor AtMYB102 Functions in Defense Against The Insect Herbivore Pieris Rapae. Plant Signal. Behav. 2006, 1, 305–311. [Google Scholar] [CrossRef]
- Ortiz-García, P.; Pérez-Alonso, M.-M.; González Ortega-Villaizán, A.; Sánchez-Parra, B.; Ludwig-Müller, J.; Wilkinson, M.D.; Pollmann, S. The Indole-3-Acetamide-Induced Arabidopsis Transcription Factor MYB74 Decreases Plant Growth and Contributes to the Control of Osmotic Stress Responses. Front. Plant Sci. 2022, 13, 928386. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, Y. A Role for Auxin in Flower Development. J. Integr. Plant Biol. 2007, 49, 99–104. [Google Scholar] [CrossRef]
- Cao, J.; Li, G.; Qu, D.; Li, X.; Wang, Y. Into the Seed: Auxin Controls Seed Development and Grain Yield. Int. J. Mol. Sci. 2020, 21, 1662. [Google Scholar] [CrossRef]
- Diener, A.C.; Gaxiola, R.A.; Fink, G.R. Arabidopsis ALF5, a Multidrug Efflux Transporter Gene Family Member, Confers Resistance to Toxins. Plant Cell 2001, 13, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Z.; Pandey, G.K.; Tsuchiya, T.; Luan, S. Functional Cloning and Characterization of a Plant Efflux Carrier for Multidrug and Heavy Metal Detoxification*. J. Biol. Chem. 2002, 277, 5360–5368. [Google Scholar] [CrossRef]
- Alengebawy, A.; Abdelkhalek, S.T.; Qureshi, S.R.; Wang, M.-Q. Heavy Metals and Pesticides Toxicity in Agricultural Soil and Plants: Ecological Risks and Human Health Implications. Toxics 2021, 9, 42. [Google Scholar] [CrossRef]
- Rose, J.K.C.; Braam, J.; Fry, S.C.; Nishitani, K. The XTH Family of Enzymes Involved in Xyloglucan Endotransglucosylation and Endohydrolysis: Current Perspectives and a New Unifying Nomenclature. Plant Cell Physiol. 2002, 43, 1421–1435. [Google Scholar] [CrossRef]
- Ishida, K.; Yokoyama, R. Reconsidering the Function of the Xyloglucan Endotransglucosylase/Hydrolase Family. J. Plant Res. 2022, 135, 145–156. [Google Scholar] [CrossRef]
- Shintani, M.; Tamura, K.; Bono, H. Meta-Analysis of Public RNA Sequencing Data of Abscisic Acid-Related Abiotic Stresses in Arabidopsis thaliana. Front. Plant Sci. 2024, 15, 1343787. [Google Scholar] [CrossRef]
- Tamura, K.; Bono, H. Meta-Analysis of RNA Sequencing Data of Arabidopsis and Rice under Hypoxia. Life 2022, 12, 1079. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Stable ID | Gene Name | Description |
|---|---|---|
| AT1G10400 | AT1G10400 | UDP-glycosyltransferase superfamily protein |
| AT1G10810 | AT1G10810 | NAD(P)-linked oxidoreductase superfamily protein |
| AT1G23800 | ALDH2B7 | aldehyde dehydrogenase 2B7 |
| AT1G60680 | AT1G60680 | NAD(P)-linked oxidoreductase superfamily protein |
| AT1G60690 | AT1G60690 | NAD(P)-linked oxidoreductase superfamily protein |
| AT1G60710 | ATB2 | NAD(P)-linked oxidoreductase superfamily protein |
| AT1G60730 | AT1G60730 | NAD(P)-linked oxidoreductase superfamily protein |
| AT1G60750 | AT1G60750 | NAD(P)-linked oxidoreductase superfamily protein |
| AT2G16890 | AT2G16890 | UDP-glycosyltransferase superfamily protein |
| AT2G46370 | JAR1 | Auxin-responsive GH3 family protein |
| AT2G46680 | HB-7 | Homeobox 7 |
| AT3G01420 | DOX1 | Peroxidase superfamily protein |
| AT3G61890 | HB-12 | Homeobox 12 |
| AT4G05100 | MYB74 | MYB domain protein 74 |
| AT4G10310 | HKT1 | High-affinity K + transporter 1 |
| AT4G21440 | MYB102 | MYB-like 102 |
| AT5G14860 | AT5G14860 | UDP-glycosyltransferase superfamily protein |
| AT5G25610 | RD22 | BURP domain-containing protein |
| Gene Stable ID | Gene Name | Description |
|---|---|---|
| AT1G04240 | SHY2 | AUX/IAA transcriptional regulator family protein |
| AT1G04250 | AXR3 | AUX/IAA transcriptional regulator family protein |
| AT1G10960 | FD1 | Ferredoxin 1 |
| AT1G20930 | CDKB2;2 | Cyclin-dependent kinase B2;2 |
| AT1G60950 | FD2 | 2Fe-2S ferredoxin-like superfamily protein |
| AT1G64590 | AT1G64590 | NAD(P)-binding Rossmann-fold superfamily protein |
| AT1G64820 | AT1G64820 | MATE efflux family protein |
| AT1G66760 | AT1G66760 | MATE efflux family protein |
| AT1G66780 | AT1G66780 | MATE efflux family protein |
| AT1G76540 | CDKB2;1 | Cyclin-dependent kinase B2;1 |
| AT2G04040 | DTX1 | MATE efflux family protein |
| AT2G04050 | AT2G04050 | MATE efflux family protein |
| AT2G04066 | AT2G04066 | MATE efflux family protein |
| AT2G04070 | AT2G04070 | MATE efflux family protein |
| AT2G04080 | AT2G04080 | MATE efflux family protein |
| AT2G04090 | AT2G04090 | MATE efflux family protein |
| AT2G04100 | AT2G04100 | MATE efflux family protein |
| AT2G13610 | ABCG5 | ABC-2 type transporter family protein |
| AT2G16850 | PIP2;8 | Plasma membrane intrinsic protein 2;8 |
| AT2G36870 | XTH32 | Xyloglucan endotransglucosylase/hydrolase 32 |
| AT2G42380 | BZIP34 | Basic-leucine zipper (bZIP) transcription factor family protein |
| AT2G48020 | AT2G48020 | Major facilitator superfamily protein |
| AT3G23030 | IAA2 | Indole-3-acetic acid inducible 2 |
| AT3G23050 | IAA7 | Indole-3-acetic acid 7 |
| AT3G58120 | BZIP61 | Basic-leucine zipper (bZIP) transcription factor family protein |
| AT4G14550 | IAA14 | Indole-3-acetic acid inducible 14 |
| AT4G14560 | IAA1 | Indole-3-acetic acid inducible |
| AT4G17340 | TIP2;2 | Tonoplast intrinsic protein 2;2 |
| AT4G25000 | AMY1 | Alpha-amylase-like protein |
| AT4G35100 | PIP3 | Plasma membrane intrinsic protein 3 |
| AT5G10130 | AT5G10130 | Pollen Ole e 1 allergen and extension family protein |
| AT5G10180 | SULTR2;1 | Sulfate transporter 2;1 |
| AT5G10570 | AT5G10570 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| AT5G43700 | ATAUX2-11 | AUX/IAA transcriptional regulator family protein |
| AT5G47450 | TIP2;3 | Tonoplast intrinsic protein 2;3 |
| AT5G65640 | bHLH093 | Beta HLH protein 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yumiya, M.; Bono, H. Meta-Analysis of Wild Relatives and Domesticated Species of Rice, Tomato, and Soybean Using Publicly Available Transcriptome Data. Life 2025, 15, 1088. https://doi.org/10.3390/life15071088
Yumiya M, Bono H. Meta-Analysis of Wild Relatives and Domesticated Species of Rice, Tomato, and Soybean Using Publicly Available Transcriptome Data. Life. 2025; 15(7):1088. https://doi.org/10.3390/life15071088
Chicago/Turabian StyleYumiya, Makoto, and Hidemasa Bono. 2025. "Meta-Analysis of Wild Relatives and Domesticated Species of Rice, Tomato, and Soybean Using Publicly Available Transcriptome Data" Life 15, no. 7: 1088. https://doi.org/10.3390/life15071088
APA StyleYumiya, M., & Bono, H. (2025). Meta-Analysis of Wild Relatives and Domesticated Species of Rice, Tomato, and Soybean Using Publicly Available Transcriptome Data. Life, 15(7), 1088. https://doi.org/10.3390/life15071088