
Computational Insight into the Rope-Skipping Isomerization of Diarylether Cyclophanes

 , and
, and
Abstract
:1. Introduction
2. Methods
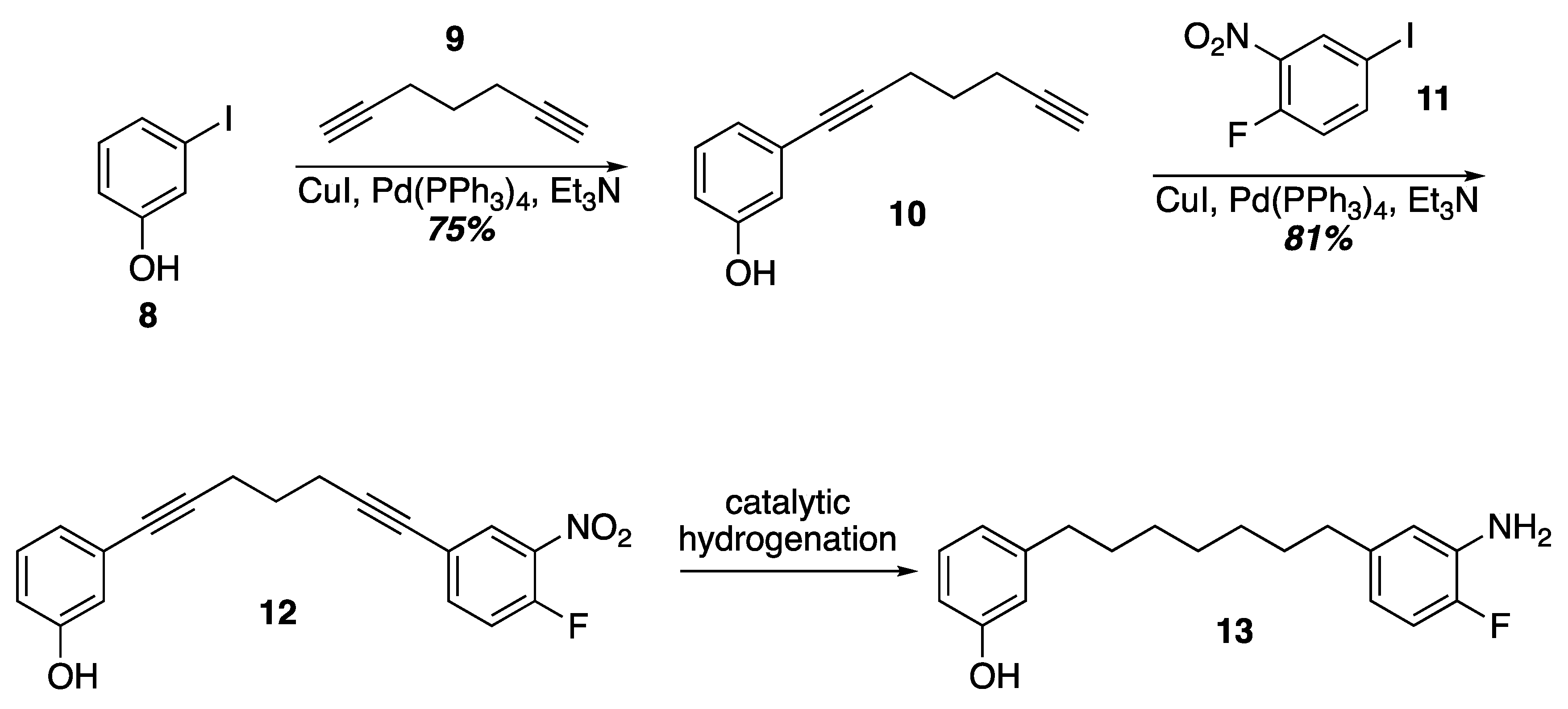
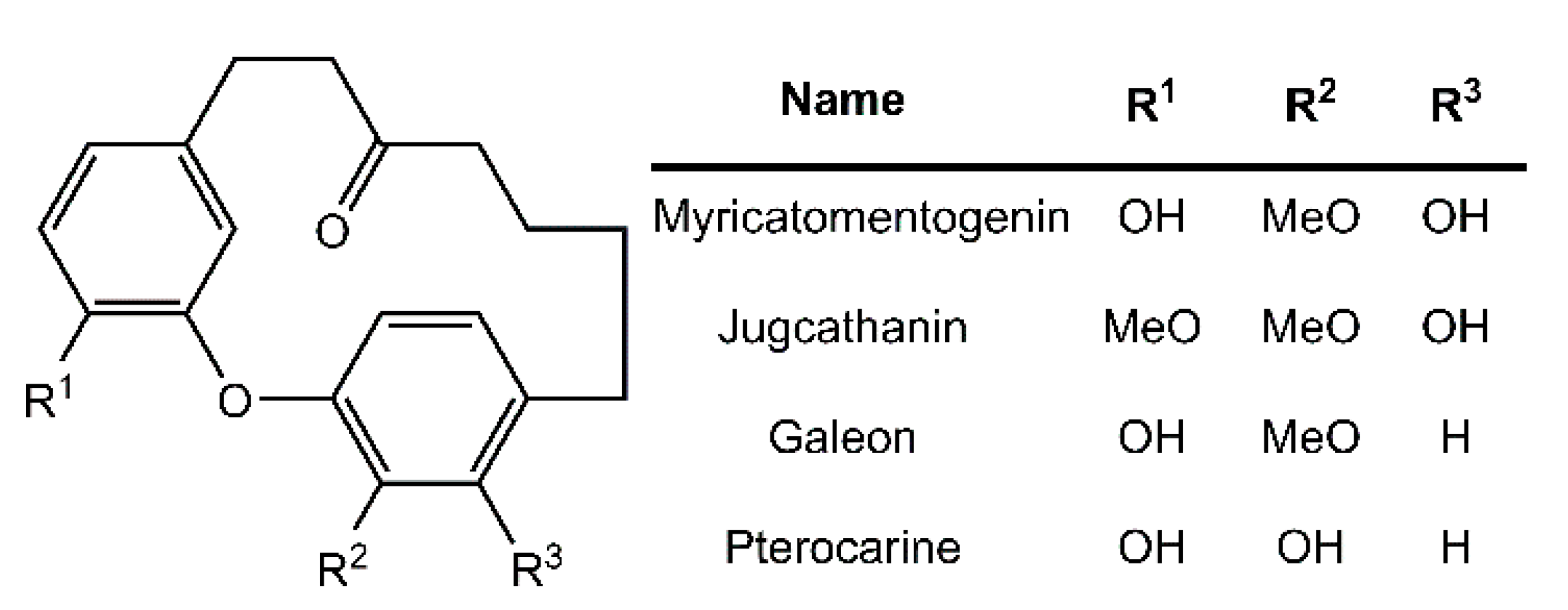
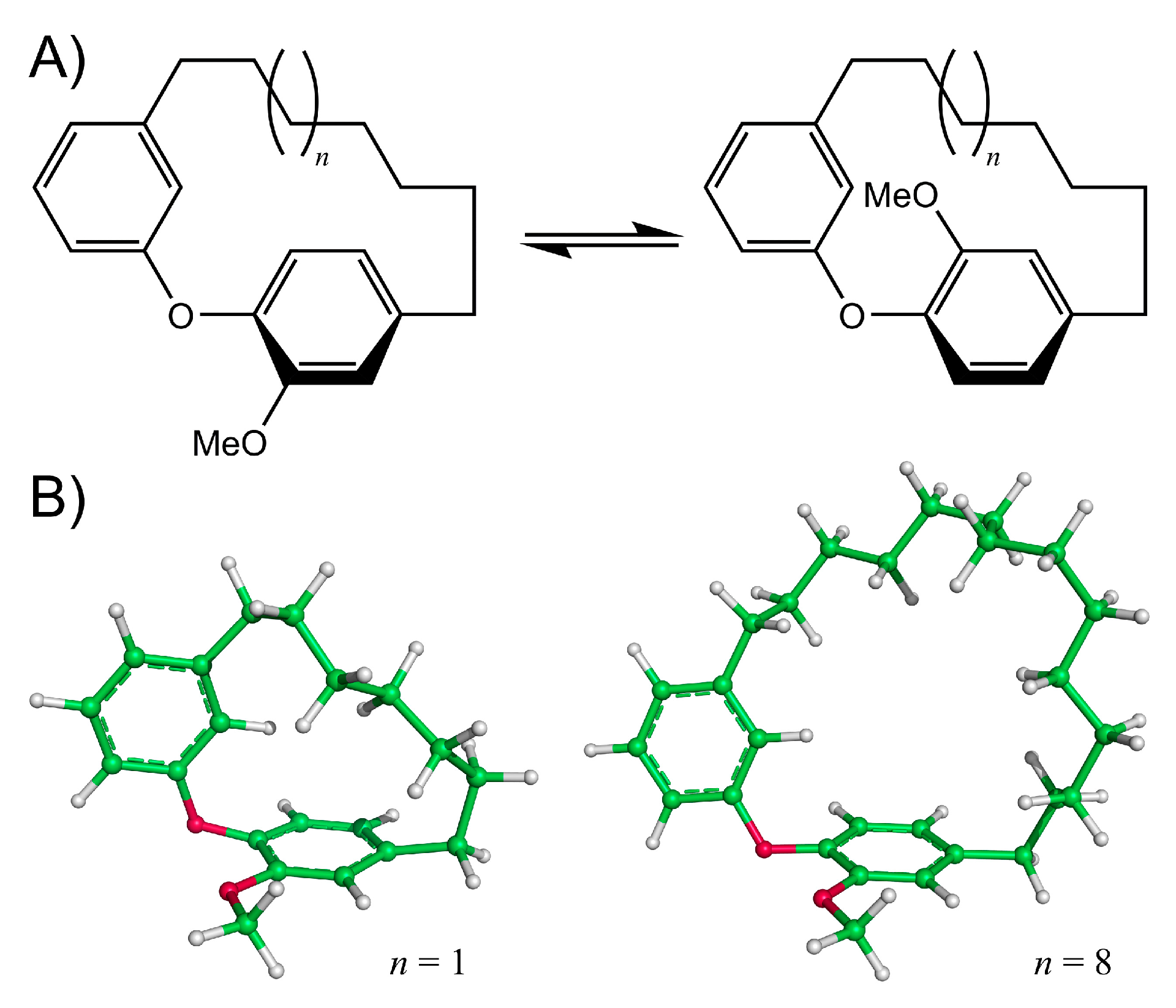
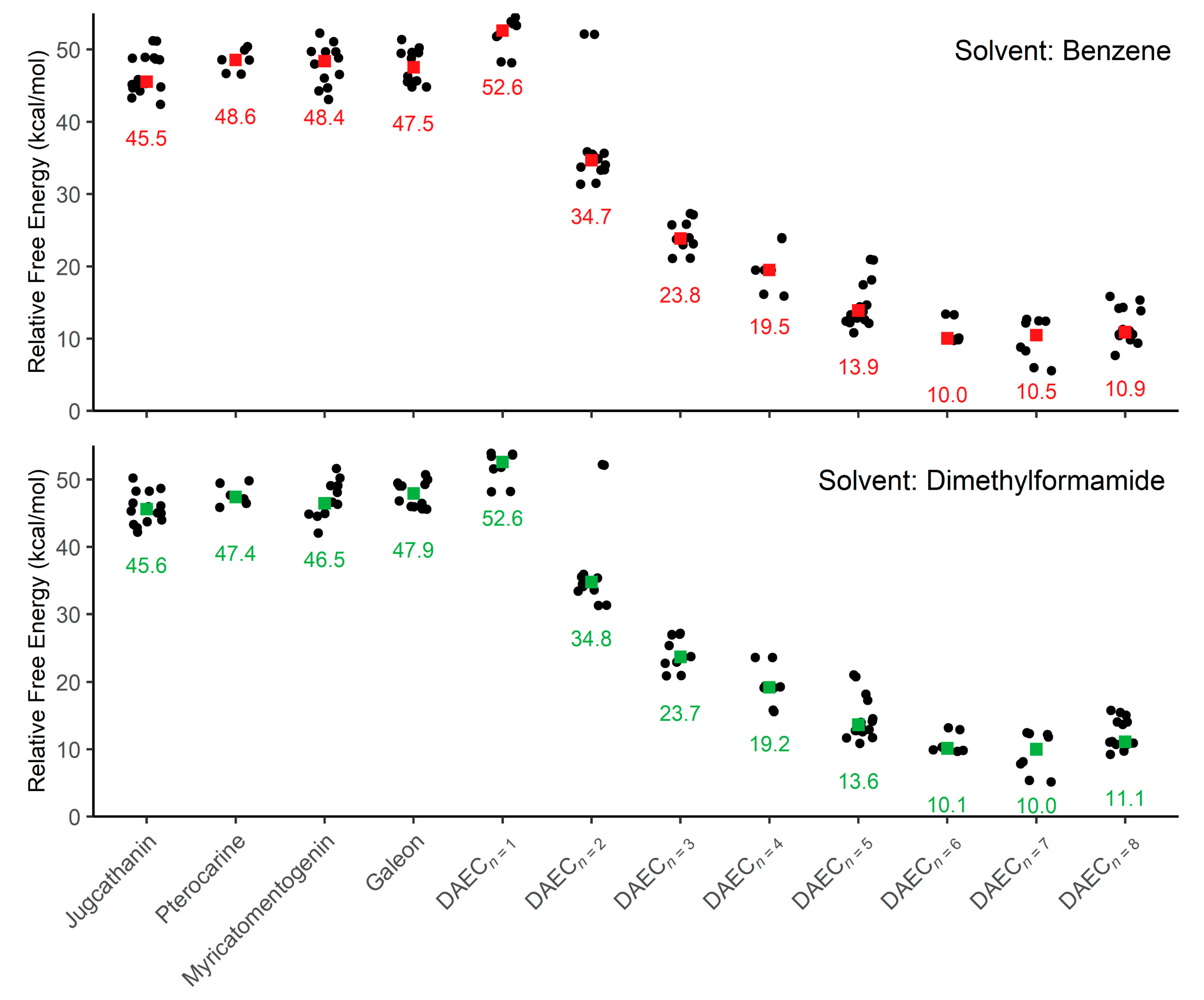
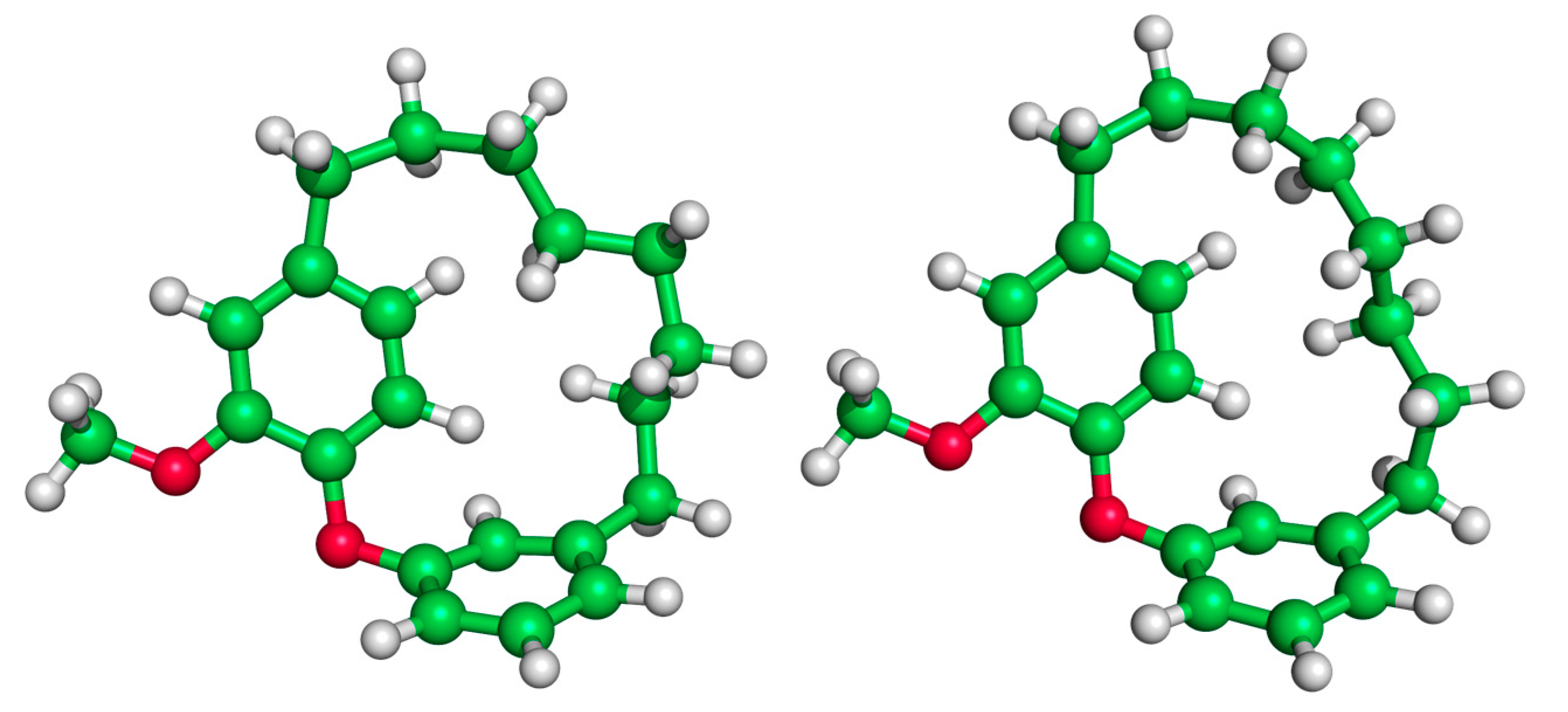
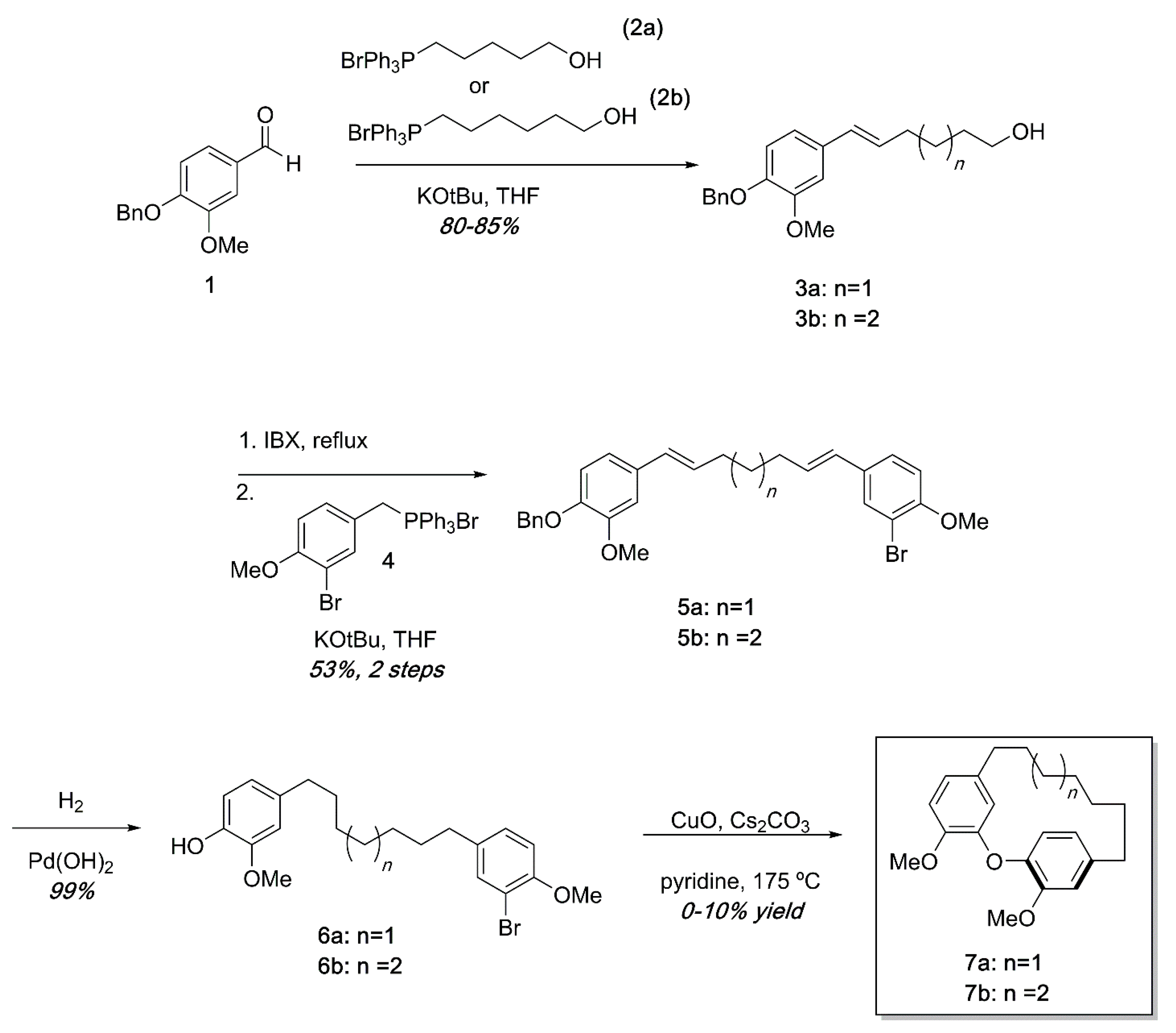
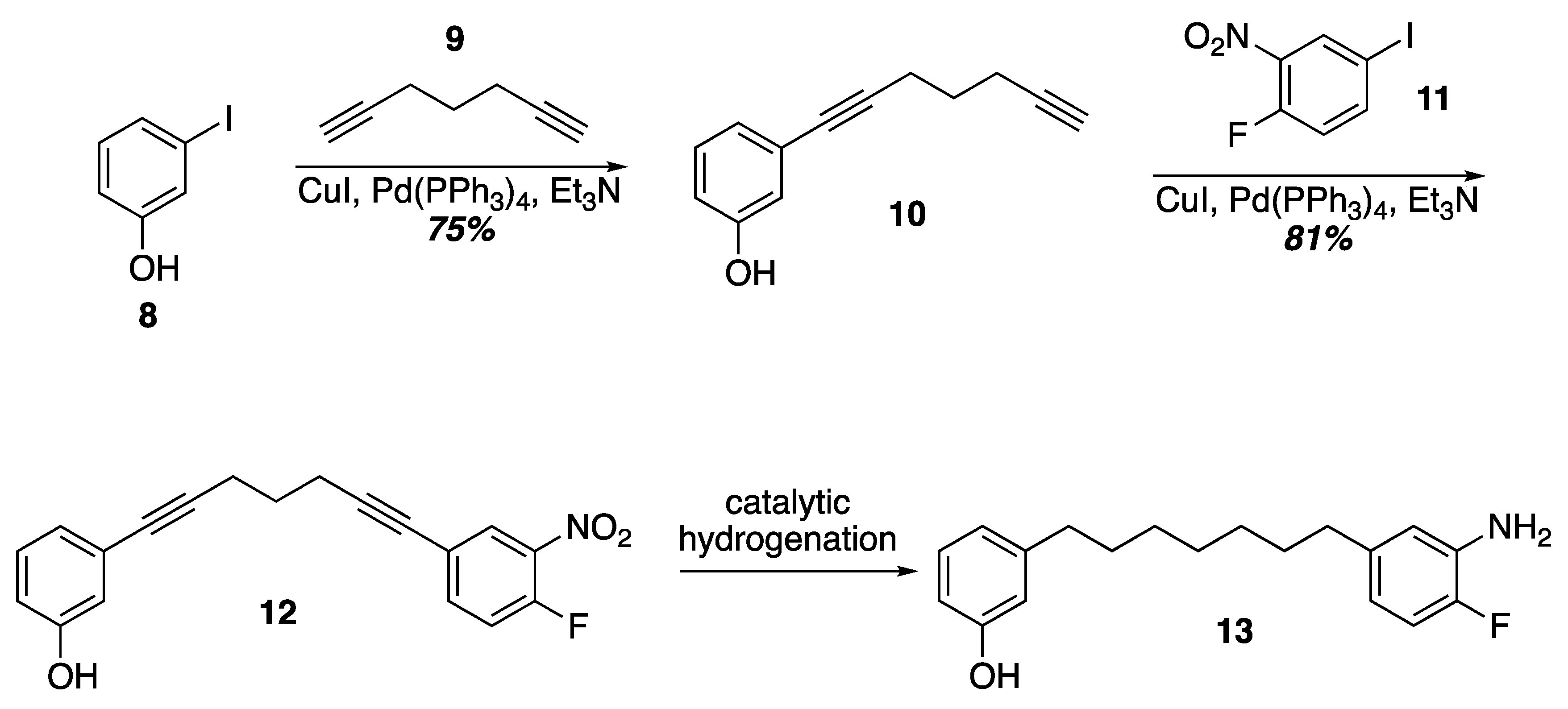
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Ōki, M. Recent Advances in Atropisomerism. In Topics in Stereochemistry; John Wiley & Sons: Hoboken, NJ, USA, 1983. [Google Scholar] [CrossRef]
- Glunz, P.W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 53–60. [Google Scholar] [CrossRef] [PubMed]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing Atropisomer Axial Chirality in Drug Discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature—The significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev. 2010, 111, 563–639. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, H.; Fujita, Y.; Banno, N.; Watanabe, K.; Kimura, Y.; Manosroi, A.; Manosroi, J.; Akihisa, T. Three new cyclic diarylheptanoids and other phenolic compounds from the bark of Myrica rubra and their melanogenesis inhibitory and radical scavenging activities. J. Oleo Sci. 2010, 59, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, M.Q.; Beaudry, C.M. Chirality in Diarylether Heptanoids: Synthesis of Myricatomentogenin, Jugcathanin and Congeners. Org. Lett. 2012, 14, 4026–4029. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Morikawa, T.; Toguchida, I.; Ando, S.; Matsuda, H.; Yoshikawa, M. Inhibitors of nitric oxide production from the bark of Myrica rubra: Structures of new biphenyl type diarylheptanoid glycosides and taraxerane type triterpene. Bioorg. Med. Chem. 2002, 10, 4005–4012. [Google Scholar] [CrossRef]
- Hochmuth, D.H.; König, W.A. Synthesis, resolution and determination of energy barriers to rotation of atropisomeric, planar-chiral [n]paracyclophanes by dynamic enantioselective gas chromatography and computer simulation. Tetrahedron Asymmetry 1999, 10, 1089–1097. [Google Scholar] [CrossRef]
- Kotha, S.; Shirbhate, M.E.; Waghule, G.T. Selected synthetic strategies to cyclophanes. Beilstein J. Org. Chem. 2015, 11, 1274–1331. [Google Scholar] [CrossRef]
- Ueda, T.; Kanomata, N.; Machida, H. Synthesis of Planar-Chiral Paracyclophanes via Samarium (II)-Catalyzed Intramolecular Pinacol Coupling. Org. Lett. 2005, 7, 2365–2368. [Google Scholar] [CrossRef] [PubMed]
- Kanda, K.; Hamanaka, R.; Endo, K.; Shibata, T. Asymmetric ortho-lithiation of 1,n-dioxa[n]paracyclophane derivatives for the generation of planar chirality. Tetrahedron 2012, 68, 1407–1416. [Google Scholar] [CrossRef]
- Scharwächter, K.P.; Hochmuth, D.H.; Dittmann, H.; König, W.A. Synthesis, resolution, and investigation of the rotational interconversion process of atropisomeric 1,n-diaza[n]paracyclophanes using cyclodextrin-mediated capillary zone electrophoresis. Chirality 2001, 13, 679–690. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Raber, D.J. A new approach to probing conformational space with molecular mechanics: Random incremental pulse search. J. Am. Chem. Soc. 1989, 111, 4371–4378. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2021. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pattawong, O.; Salih, M.Q.; Rosson, N.T.; Beaudry, C.M.; Cheong, P.H.-Y. The nature of persistent conformational chirality, racemization mechanisms, and predictions in diarylether heptanoid cyclophane natural products. Org. Biomol. Chem. 2014, 12, 3303–3309. [Google Scholar] [CrossRef]
- Zhu, Z.-Q.; Salih, M.Q.; Fynn, E.; Bain, A.D.; Beaudry, C.M. The Garuganin and Garugamblin Diarylether Heptanoids: Total Synthesis and Determination of Chiral Properties Using Dynamic NMR. J. Org. Chem. 2013, 78, 2881–2896. [Google Scholar] [CrossRef]
- Gonzalez, G.I.; Zhu, J. A Unified Strategy toward the Synthesis of Acerogenin-Type Macrocycles: Total Syntheses of Acerogenins A, B, C, and L and Aceroside IV. J. Org. Chem. 1999, 64, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Atkinson, J. Synthesis of Phytyl- and Chroman-Derivatized Photoaffinity Labels Based on α-Tocopherol. J. Org. Chem. 2000, 65, 2560–2567. [Google Scholar] [CrossRef] [PubMed]
- More, J.D.; Finney, N.S. A Simple and Advantageous Protocol for the Oxidation of Alcohols with o-Iodoxybenzoic Acid (IBX). Org. Lett. 2002, 4, 3001–3003. [Google Scholar] [CrossRef] [PubMed]
- Burgos, C.H.; Barder, T.E.; Huang, X.; Buchwald, S.L. Significantly Improved Method for the Pd-Catalyzed Coupling of Phenols with Aryl Halides: Understanding Ligand Effects. Angew. Chem. Int. Ed. 2006, 45, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Jourdant, A.; González-Zamora, E.; Zhu, J. Wilkinson’s Catalyst Catalyzed Selective Hydrogenation of Olefin in the Presence of an Aromatic Nitro Function: A Remarkable Solvent Effect. J. Org. Chem. 2002, 67, 3163–3164. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Ren, J.; Zeng, B.-B. Oxidation of aromatic amines into nitroarenes with m-CPBA. Tetrahedron Lett. 2014, 55, 1581–1584. [Google Scholar] [CrossRef]
- Guan, Y.; Ingman, V.M.; Rooks, B.J.; Wheeler, S.E. AARON: An Automated Reaction Optimizer for New Catalysts. J. Chem. Theory Comput. 2018, 14, 5249–5261. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Bochevarov, A.D.; Watson, M.A.; Hughes, T.F.; Rinaldo, D.; Ehrlich, S.; Steinbrecher, T.B.; Vaitheeswaran, S.; Philipp, D.M.; Halls, M.D.; et al. Automated Transition State Search and Its Application to Diverse Types of Organic Reactions. J. Chem. Theory Comput. 2017, 13, 5780–5797. [Google Scholar] [CrossRef]
- Robertson, C.; Habershon, S. Simple position and orientation preconditioning scheme for minimum energy path calculations. J. Comput. Chem. 2021, 42, 761–770. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Ono, Y.; Taketsugu, T.; Morokuma, K. Intrinsic reaction coordinate: Calculation, bifurcation, and automated search. Int. J. Quantum Chem. 2014, 115, 258–269. [Google Scholar] [CrossRef]

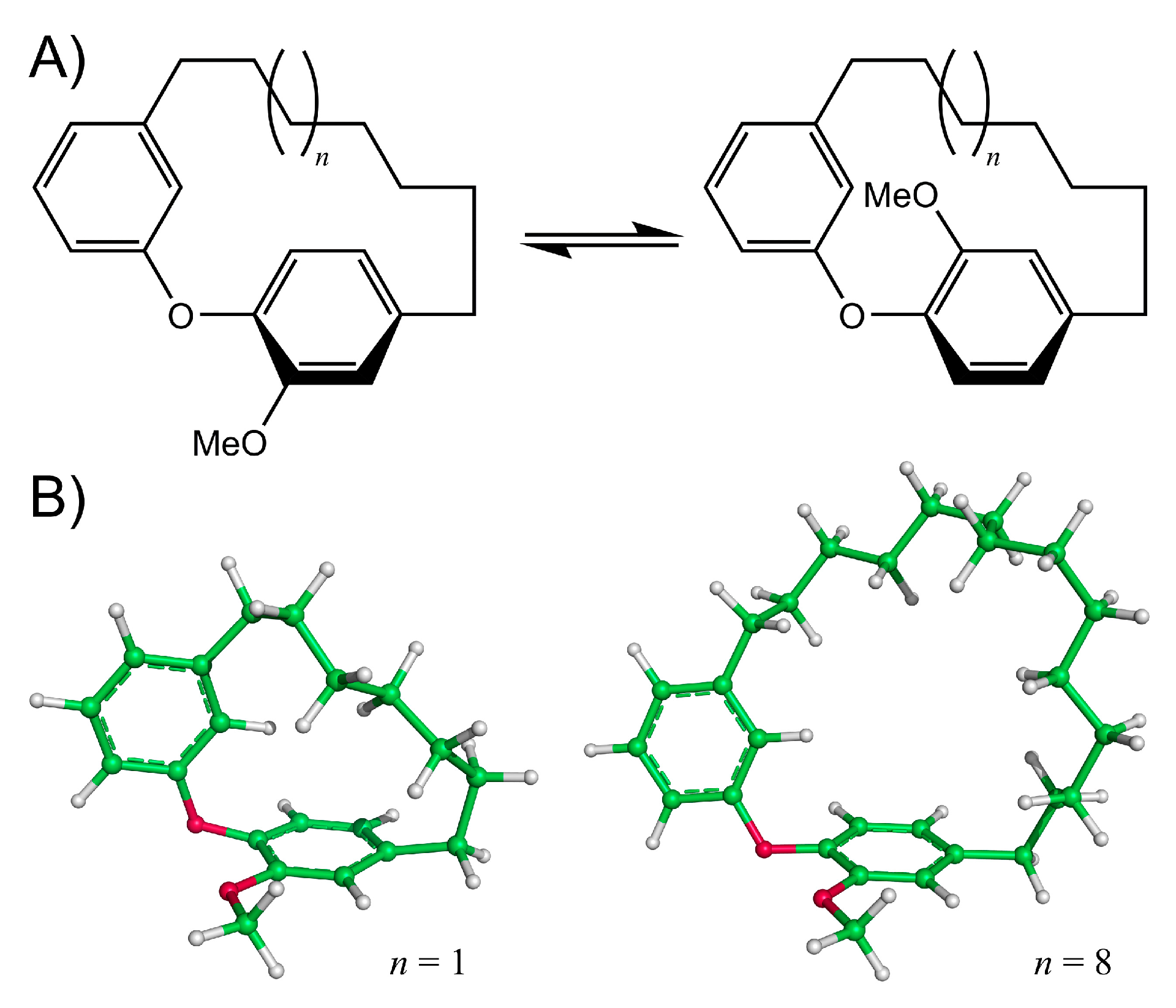
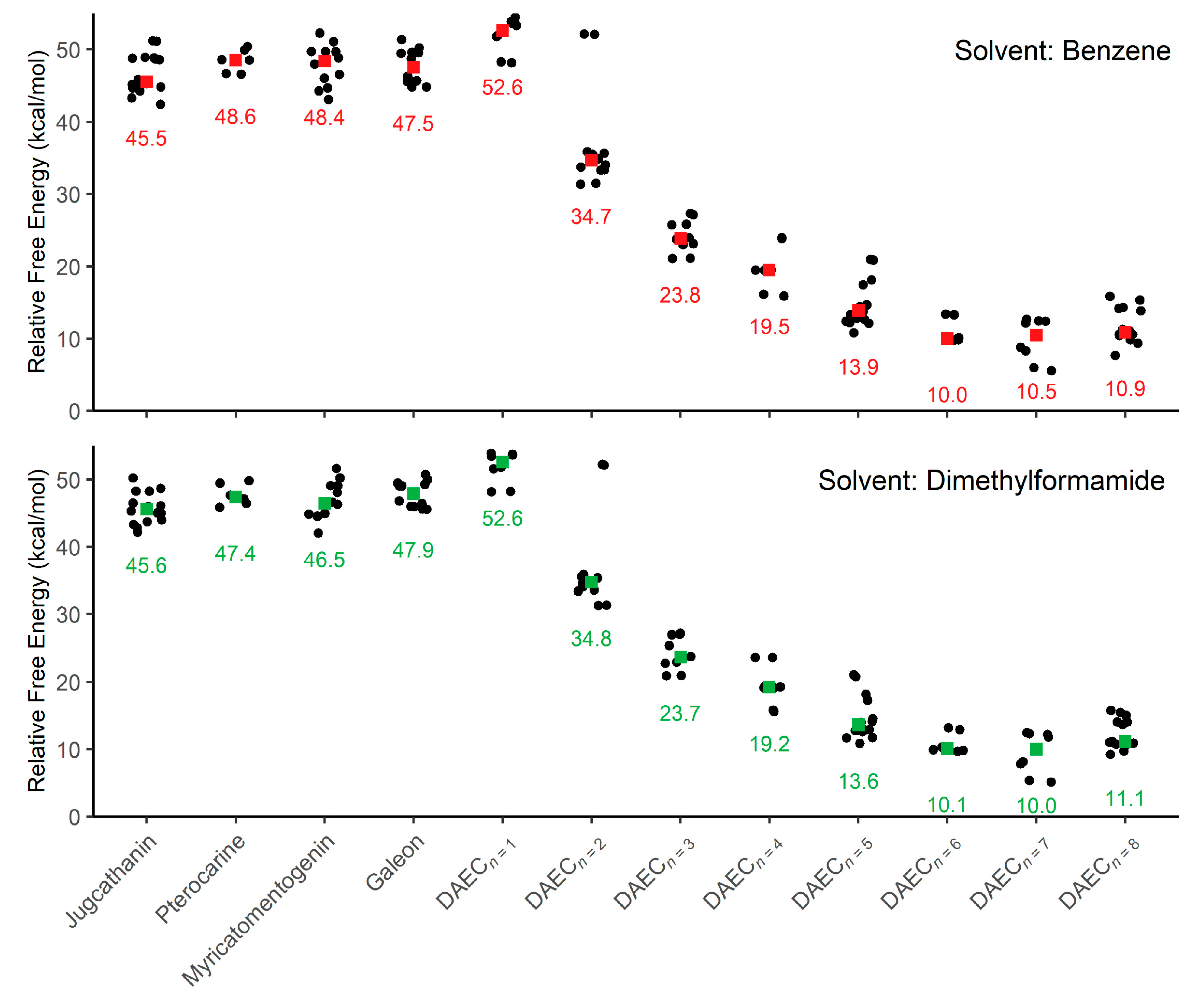
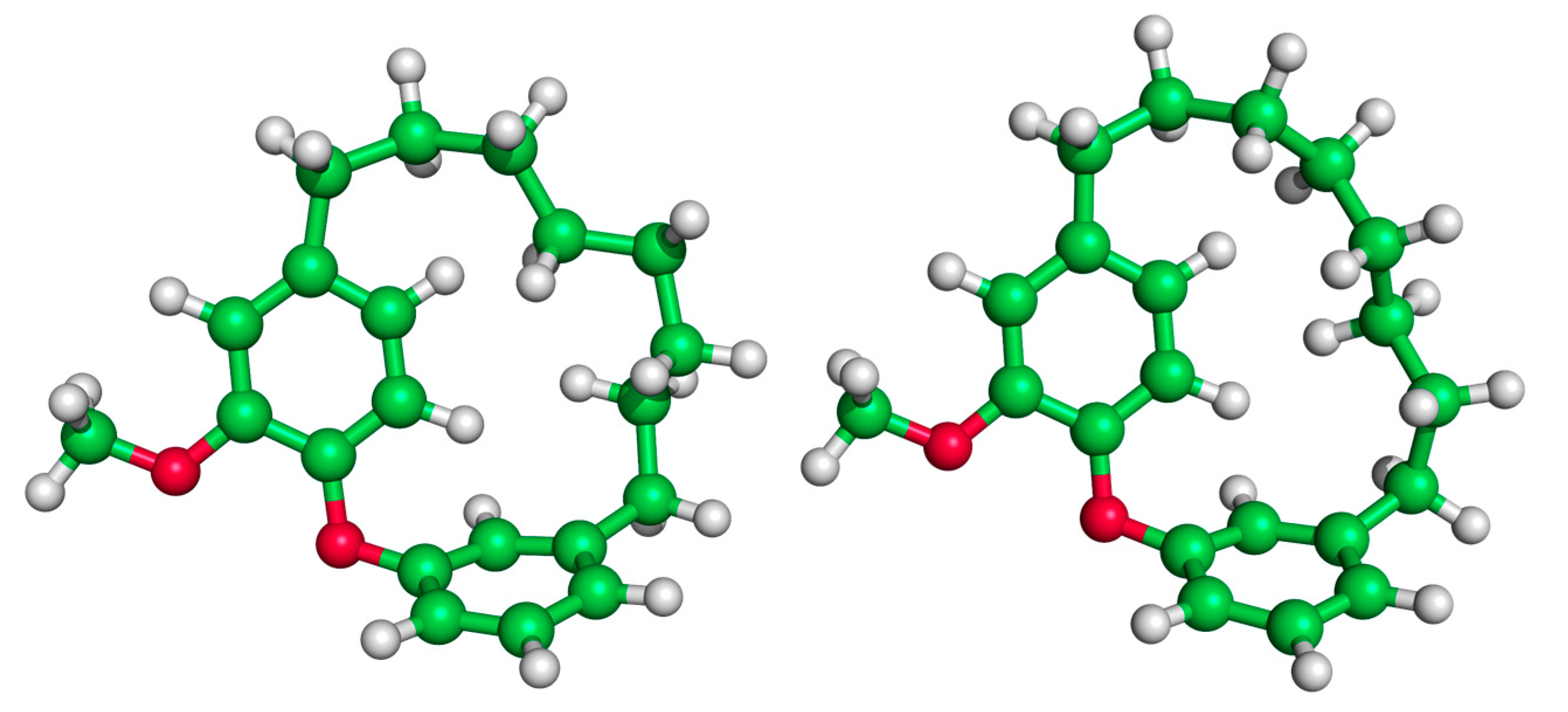
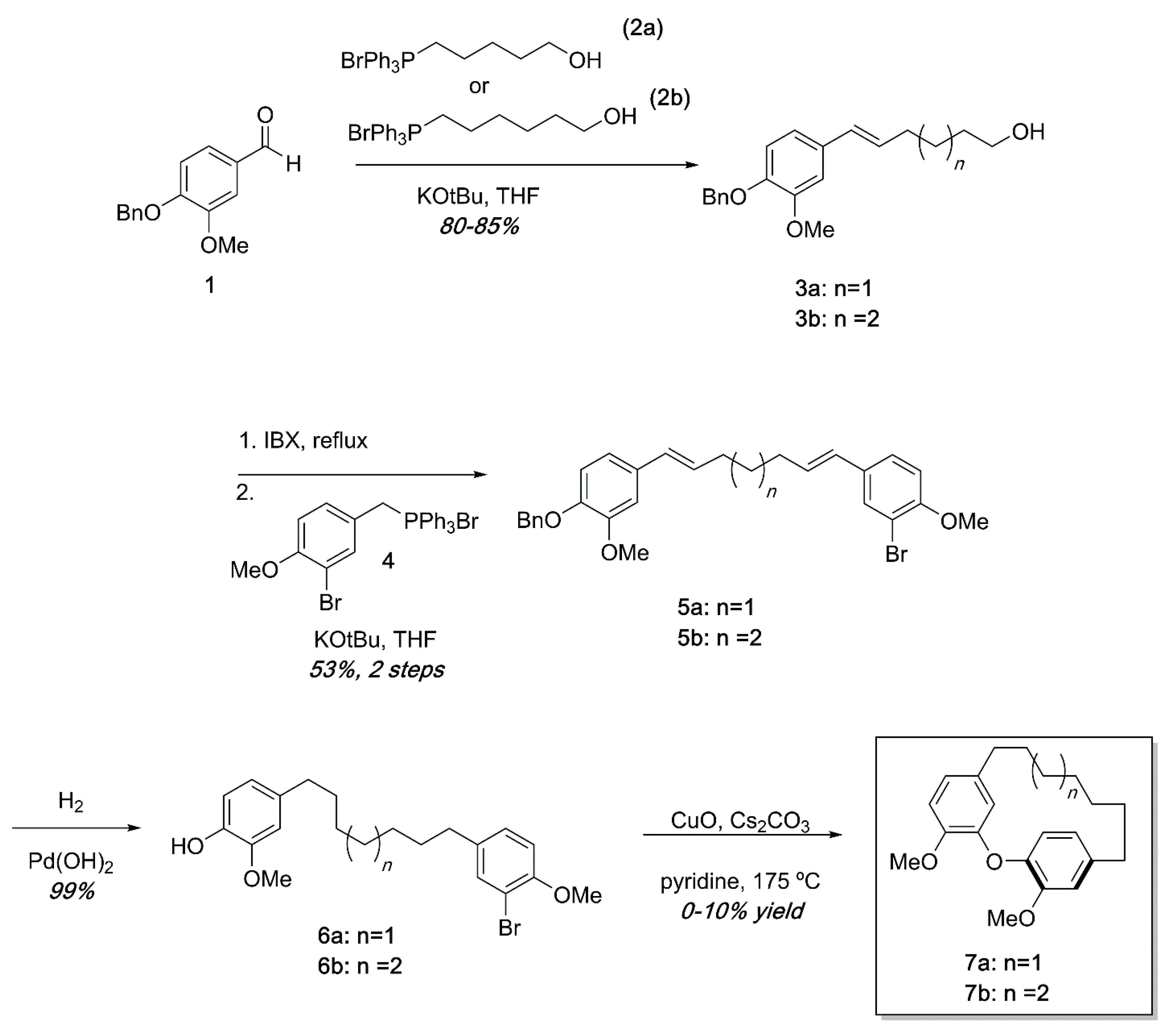

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Mole Fraction of RIPS Conformers Selected for QM Optimization | Mole Fraction of 7 Sampled QM-Optimized Models |
|---|---|---|
| Jugcathanin | 1.00 | 0.960 |
| Pterocarine | 1.00 | 0.993 |
| Myricatomentogenin | 1.00 | 0.959 |
| Galeon | 1.00 | 0.957 |
| DAECn=1 | 1.00 | 0.720 |
| DAECn=2 | 0.999 | 0.585 |
| DAECn=3 | 0.998 | 0.395 |
| DAECn=4 | 0.997 | 0.379 |
| DAECn=5 | 0.985 | 0.297 |
| DAECn=6 | 0.981 | 0.471 |
| DAECn=7 | 0.604 | -- a |
| DAECn=8 | 0.606 | -- a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Summers, T.J.; Tupkar, H.; Ozvat, T.M.; Tregillus, Z.; Miller, K.A.; DeYonker, N.J. Computational Insight into the Rope-Skipping Isomerization of Diarylether Cyclophanes. Symmetry 2021, 13, 2127. https://doi.org/10.3390/sym13112127
Summers TJ, Tupkar H, Ozvat TM, Tregillus Z, Miller KA, DeYonker NJ. Computational Insight into the Rope-Skipping Isomerization of Diarylether Cyclophanes. Symmetry. 2021; 13(11):2127. https://doi.org/10.3390/sym13112127
Chicago/Turabian StyleSummers, Thomas J., Hrishikesh Tupkar, Tyler M. Ozvat, Zoë Tregillus, Kenneth A. Miller, and Nathan J. DeYonker. 2021. "Computational Insight into the Rope-Skipping Isomerization of Diarylether Cyclophanes" Symmetry 13, no. 11: 2127. https://doi.org/10.3390/sym13112127
APA StyleSummers, T. J., Tupkar, H., Ozvat, T. M., Tregillus, Z., Miller, K. A., & DeYonker, N. J. (2021). Computational Insight into the Rope-Skipping Isomerization of Diarylether Cyclophanes. Symmetry, 13(11), 2127. https://doi.org/10.3390/sym13112127