
Investigation of Pharmaceutical Importance of 2H-Pyran-2-One Analogues via Computational Approaches

 ,
,  ,
,  ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
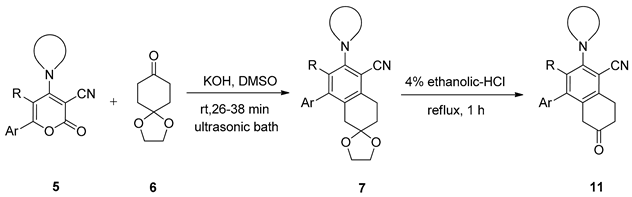
2.1. General Procedure for the Synthesis of Functionalized Spirocyclic Ketals 7
2.2. General Procedure for the Synthesis of Functionalized 2-Tetralone 11
2.3. Computational Details
3. Results and Discussions
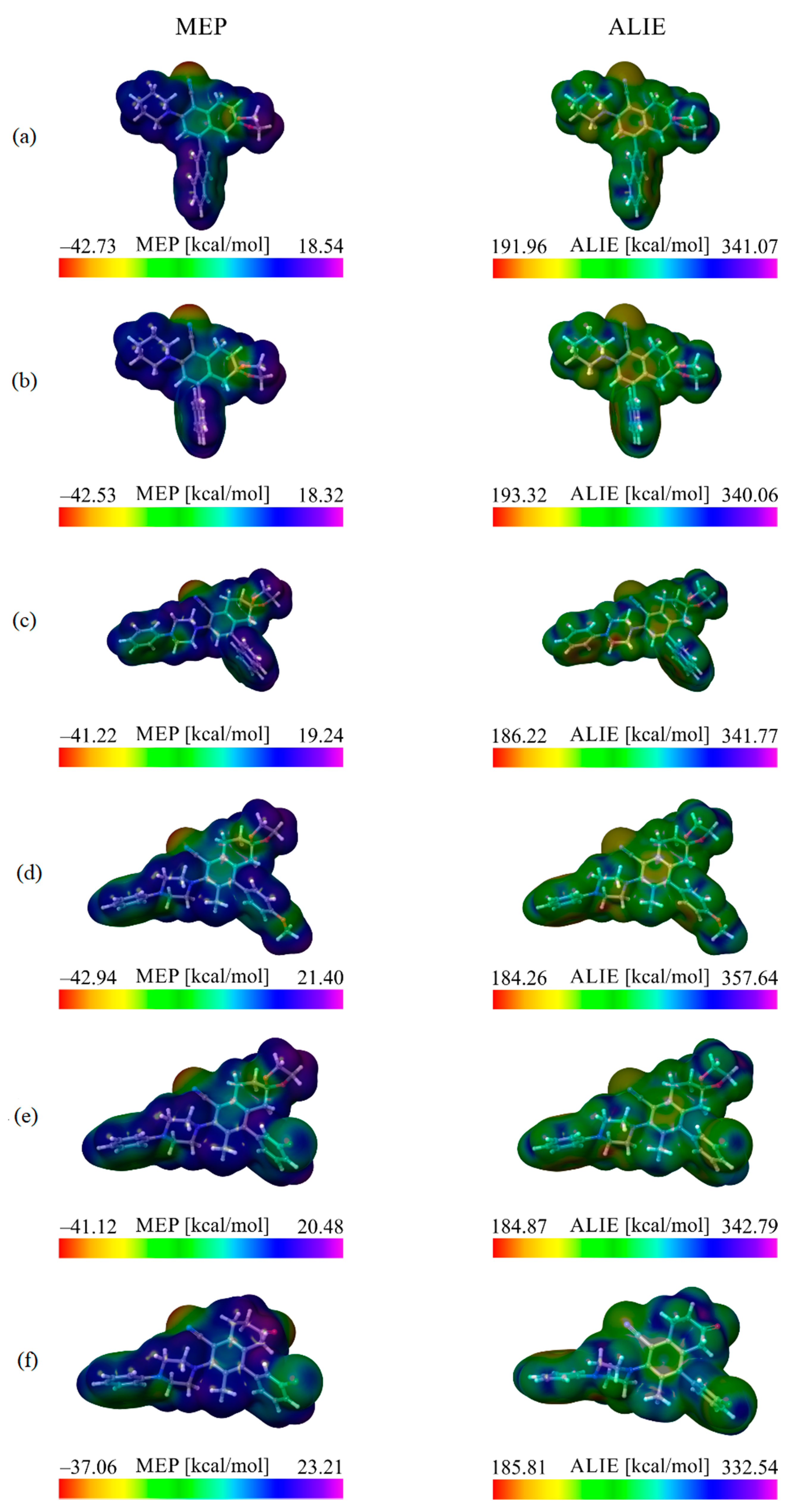
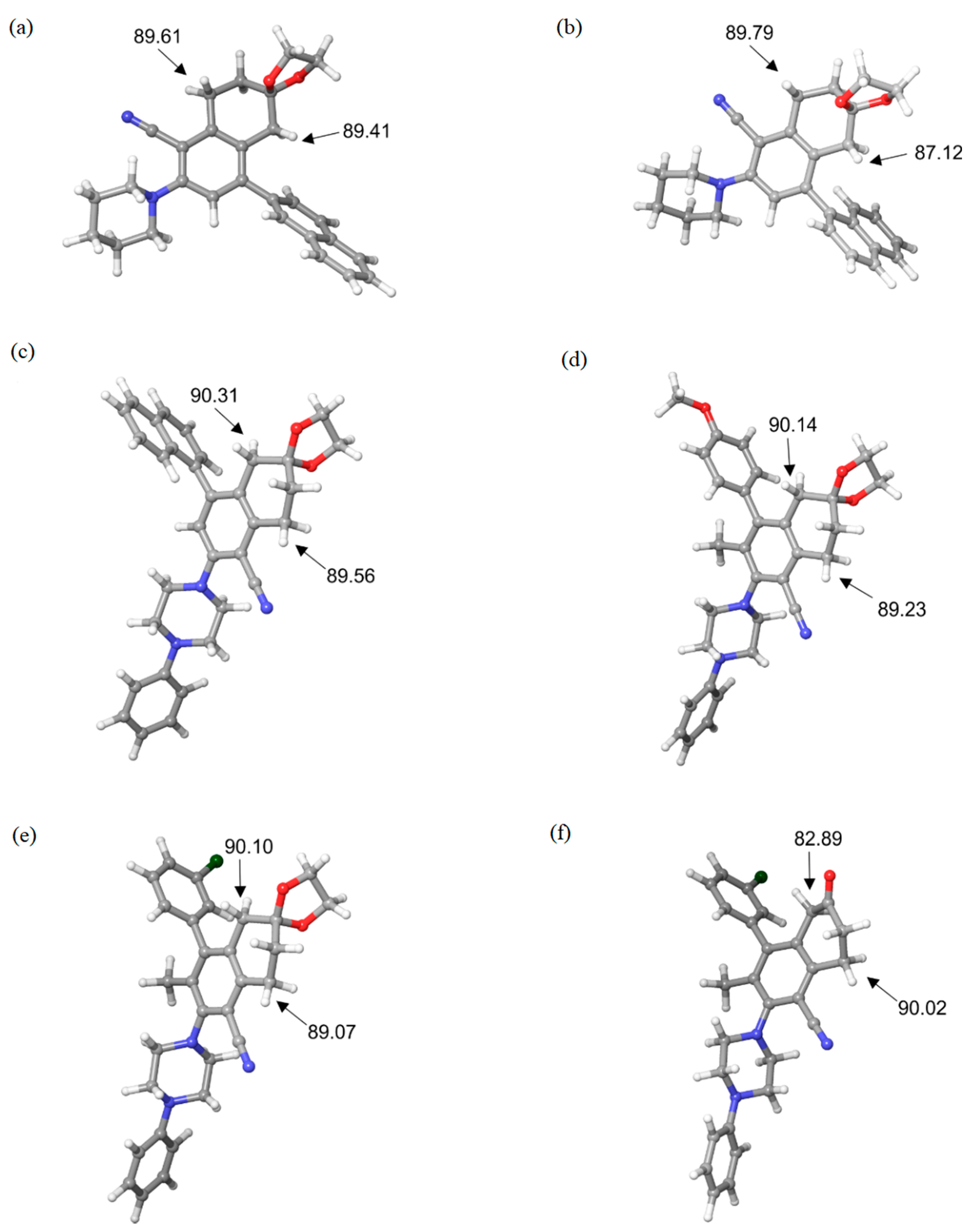
3.1. Local Reactivity Properties–MEP and ALIE Surfaces
3.2. Sensitivity towards Autoxidation
3.3. Interactions with Water
3.4. Identification of Suitable Excipients
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silveira, C.C.; Braga, A.; Kaufman, T.; Lenardao, E.J. Synthetic approaches to 2-tetralones. Tetrahedron 2004, 60, 8295–8328. [Google Scholar] [CrossRef]
- Makhey, D.; Li, D.; Zhao, B.; Sim, S.-P.; Li, T.-K.; Liu, A.; Liu, L.F.; LaVoie, E.J. Substituted Benzo[i]phenanthridines as Mammalian Topoisomerase-Targeting Agents. Bioorg. Med. Chem. 2003, 11, 1809–1820. [Google Scholar] [CrossRef]
- El Sayed, K.A.; Foudah, A.I.; Mayer, A.M.S.; Crider, A.M.; Song, D. Synthesis, microbial transformation, and pharmacological evaluation of 4,5-dihydronaphtho[2,1-b]furan-2-ones and related analogues. MedChemComm 2013, 4, 1231–1238. [Google Scholar] [CrossRef]
- Pautigny, C.; Debouit, C.; Vayron, P.; Ayad, T.; Ratovelomanana-Vidal, V. Ratovelomanana-Vidal, Asymmetric hydrogenation of trisubstituted-N-acetyl enamides derived from 2-tetralones using ruthenium-SYNPHOS catalysts: A practical synthetic approach to the preparation of β3-adrenergic agonist SR58611A. Tetrahedron Asymmetry 2010, 21, 1382–1388. [Google Scholar] [CrossRef]
- Balijapalli, U.; Udayadasan, S.; Shanmugam, E.; Iyer, S.K. Synthesis, photophysical and acidochromic properties of a series of tetrahydrodibenzo[a,i]phenanthridine chromophores. Dye Pigment. 2016, 130, 233–244. [Google Scholar] [CrossRef]
- Umamahesh, B.; Sathiskumar, U.; Augustine, G.; Easwaramoorthi, S.; Ayyadurai, N.; Sathiyanarayanan, K.I. Photophysical studies of donor, acceptor substituted tetrahydrodibenzo[a,i]phenanthridines. Dye. Pigment. 2016, 134, 409–418. [Google Scholar] [CrossRef]
- Ponram, M.; Balijapalli, U.; Sambath, B.; Iyer, S.K.; Venkatachalapathy, B.; Cingaram, R.; Sundaramurthy, K.N. Development of paper-based chemosensor for the detection of mercury ions using mono- and tetra-sulfur bearing phenanthridines. New J. Chem. 2018, 42, 8530–8536. [Google Scholar] [CrossRef]
- Muthukumar, V.; Iyer, S.K. Highly selective chemosensor for the detection of Ru3+ ion byfluorescent turn-on response and its bioimagingrecognitionin living cells. Sens. Actuators B 2018, 267, 373–380. [Google Scholar]
- Goel, A.; Sharma, A.; Rawat, M.; Anand, R.S.; Kant, R. Synthesis of Fluorescent C2-Bridged Teraryls and Quateraryls for Blue, Sky-Blue, and Green Color Light-Emitting Devices. J. Org. Chem. 2014, 79, 10873–10880. [Google Scholar] [CrossRef]
- Wang, B.; Peng, F.; Huang, W.; Zhou, J.; Zhang, N.; Sheng, J.; Haruehanroengra, P.; He, G.; Han, B. Rational drug design, synthesis, and biological evaluation of novel chiral tetrahydronaphthalene-fused spirooxindole as MDM2-CDK4 dual inhibitor against glioblastoma. Acta Pharm. Sin. B 2020, 10, 1492–1510. [Google Scholar] [CrossRef]
- Ates-Alagoz, Z.; Yildiz, S.; Buyukbingol, E. Antimicrobial Activities of Some Tetrahydronaphthalene-Benzimidazole Derivatives. Chemotherapy 2007, 53, 110–113. [Google Scholar] [CrossRef]
- Uppal, A.; Kothiyal, P.; Singh, A. Hybrid Class Phenylthiazole and 1,2,3,4-Tetrahydronaphthalene Target Sertraline Transporter for Antidepressant Action Revealed by Molecular Docking Studies. J. Heterocycl. Chem. 2015, 52, 1429–1437. [Google Scholar] [CrossRef]
- Barker, M.; Clackers, M.; Copley, R.; Demaine, D.A.; Humphreys, D.; Inglis, G.G.A.; Johnston, M.J.; Jones, H.T.; Haase, M.V.; House, D.; et al. Dissociated Nonsteroidal Glucocorticoid Receptor Modulators; Discovery of the Agonist Trigger in a Tetrahydronaphthalene-Benzoxazine Series. J. Med. Chem. 2006, 49, 4216–4231. [Google Scholar] [CrossRef] [PubMed]
- Rho, Y.S.; Ko, H.K.; Sin, H.; Yoo, D.J. A Facile Total Synthesis of Idarubicinone. Bull. Kor. Chem. Soc. 1999, 20, 1517–1520. [Google Scholar]
- Hauser, F.M.; Prasanna, S. Total syntheses of (.+-.)-duanomycinone. Regiospecific preparations of (.+-.)-7,9-dideoxydaunomycinone and 6,11-dihydroxy-4-methoxy-7,8,9,10-tetrahydronaphthacene-5,9,12-trione. J. Am. Chem. Soc. 1981, 103, 6378–6386. [Google Scholar] [CrossRef]
- Franco, T.; Silvana, F. A process for the Preparation of Nepinalone. European Patent ApplEP0507001A1, 7 October 1992. [Google Scholar]
- Cobley, C.J.; Evans, G.; Fanjul, T.; Simmonds, S.; Woods, A. New catalytic route for the synthesis of an optically active tetralone-derived amine for rotigotine. Tetrahedron Lett. 2016, 57, 986–989. [Google Scholar] [CrossRef]
- Batra, H.; Tuladhar, S.M. An Improved Process to Prepare Treprostinil, the Active Ingredient in Remodulin ®. WO 2009/078965, 25 June 2009. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009078965 (accessed on 18 June 2021).
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An overview of molecular modelling for drug discovery with specific illustrative examples of applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Mary, Y.S.; Resmi, K.S.; Narayana, B.; Sarojini, S.B.K.; Armaković, S.; Armaković, S.J.; Vijayakumar, G.; van Alsenoy, C.; Mohan, B.J. Synthesis and spectroscopic study of two new pyrazole derivatives with detailed computational evaluation of their reactivity and pharmaceutical potential. J. Mol. Struct. 2019, 1181, 599–612. [Google Scholar] [CrossRef]
- Beegum, S.; Mary, Y.S.; Thomas, R.; Armaković, S.; Armaković, S.J.; Zitko, J.; Dolezal, M.; Van Alsenoy, C. Exploring the detailed spectroscopic characteristics, chemical and biological activity of two cyanopyrazine-2-carboxamide derivatives using experimental and theoretical tools. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2020, 224, 117414. [Google Scholar] [CrossRef] [PubMed]
- Zainuri, D.A.; Arshad, S.; Khalib, N.C.; Razak, I.A.; Pillai, R.R.; Sulaiman, S.F.; Hashim, N.S.; Ooi, K.L.; Armaković, S.; Armaković, S.J. Synthesis, XRD crystal structure, spectroscopic characterization (FT-IR, 1H and 13C NMR), DFT studies, chemical reactivity and bond dissociation energy studies using molecular dynamics simulations and evaluation of antimicrobial and antioxidant activities of a novel chalcone derivative,(E)-1-(4-bromophenyl)-3-(4-iodophenyl) prop-2-en-1-one. J. Mol. Struct. 2017, 1128, 520–533. [Google Scholar]
- Mary, Y.S.; Aswathy, V.V.; Panicker, C.Y.; Bielenica, A.; Brzózka, P.; Savczenko, O.; Armaković, S.; Armaković, S.J.; Van Alsenoy, C. Spectroscopic, single crystal XRD structure, DFT and molecular dynamics investigation of 1-(3-chloro-4-fluorophenyl)-3-[3-(trifluoromethyl)phenyl]thiourea. RSC Adv. 2016, 6, 111997–112015. [Google Scholar] [CrossRef]
- Pillai, R.R.; Karrouchi, K.; Fettach, S.; Armaković, S.; Armaković, S.; Brik, Y.; Taoufik, J.; Radi, S.; Faouzi, M.E.A.; Ansar, M. Synthesis, spectroscopic characterization, reactive properties by DFT calculations, molecular dynamics simulations and biological evaluation of Schiff bases tethered 1,2,4-triazole and pyrazole rings. J. Mol. Struct. 2019, 1177, 47–54. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Bochevarov, A.D.; Watson, M.A.; Hughes, T.F.; Rinaldo, D.; Ehrlich, S.; Steinbrecher, T.B.; Vaitheeswaran, S.; Philipp, D.M.; Halls, M.D.; et al. Automated Transition State Search and Its Application to Diverse Types of Organic Reactions. J. Chem. Theory Comput. 2017, 13, 5780–5797. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-3: Jaguar, Schrödinger; LLC: New York, NY, USA, 2020; Available online: https://www.schrodinger.com/products/jaguar (accessed on 18 June 2021).
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06 Proc. 2006 ACM/IEEE Conf. Supercomput, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Schrödinger Release 2020-3: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA, 2020.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. Int. Forces 1981, 331–342. [Google Scholar]
- Goel, A.; Ram, V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron 2009, 65, 7865–7913. [Google Scholar] [CrossRef]
- Pratap, R.; Ram, V.J. 2H-Pyran-2-ones and their annelatedanalogs as multifaceted building blocks for the fabrication of diverse heterocycles. Tetrahedron 2017, 73, 2529–2590. [Google Scholar] [CrossRef]
- Singh, F.V.; Kole, P.B. Metal-Free Synthesis of Biaryl- and Teraryl-Cored Diarylmethanes by Ring Transformation of 2H-Pyran-2-ones. Synthesis 2019, 51, 1435–1444. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Singh, F.V. Ultrasound-assisted one pot synthesis of polysubstituted meta-terphenyls using ring transformation strategy. Synth. Commun. 2019, 49, 1092–1102. [Google Scholar] [CrossRef]
- Kole, P.B.; Singh, F.V. Versatile Synthesis of Functionalized Tetrahydroisoquinolines by Ring Transformation of 2H-Pyran-2-ones. Aust. J. Chem. 2019, 72, 524. [Google Scholar] [CrossRef] [Green Version]
- Shetgaonkar, S.E.; Singh, F.V. Base-mediated alternate route for multi-functionalized allylbenzenes using ring transformation strategy. Synth. Commun. 2020, 50, 2511–2521. [Google Scholar] [CrossRef]
- Chandrasekar, S.; Singh, F.V. Metal-Free Synthesis of Thermally Stable Fluorescent p-Terphenyls by Ring Transformation of 2H-Pyran-2-ones. ChemistrySelect 2020, 5, 7452–7459. [Google Scholar] [CrossRef]
- Kanimozhi, R.; Singh, F.V. Metal-free synthesis and characterization of 1,3-Bis(heteroaryl)benzenes followed by the photophysical studies using ultraviolet–visible and fluorescence spectroscopy. J. Mol. Struct. 2020, 1219, 128633. [Google Scholar] [CrossRef]
- Chandrasekar, S.; Kennedy, L.J.; Singh, F.V. Synthesis, spectral characterization and photophysical studies of tetrahydroquinolines. J. Mol. Struct. 2020, 1226, 129365. [Google Scholar]
- Tominaga, Y.; Ushirogochi, A.; Matsuda, Y. Synthesis and reaction of 6-substituted 3-methoxycarbonyl-4-methylthio-2H-pyran-2-one derivatives. J. Heterocycl. Chem. 1987, 24, 1557–1567. [Google Scholar] [CrossRef]
- Tominaga, Y. Synthesis of heterocyclic compounds using ketene dithioacetals. Trends Heterocycl. Chem. 1991, 2, 43. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Singh, F.V. A Metal-Free Approach for the Synthesis of 2-Tetralones via Carbanion-Induced Ring Transformation of 2H-Pyran-2-ones. Synthesis 2018, 50, 3540–3548. [Google Scholar]
- Andersson, T.; Broo, A.; Evertsson, E. Prediction of drug candidates’ sensitivity toward autoxidation: Computational estimation of CH dissociation energies of carbon-centered radicals. J. Pharm. Sci. 2014, 103, 1949–1955. [Google Scholar] [CrossRef]
- Raillard, S.P.; Bercu, J.; Baertschi, S.; Riley, C.M. Prediction of Drug Degradation Pathways leading to Structural Alerts for Potential Genotoxic Impurities. Org. Process. Res. Dev. 2010, 14, 1015–1020. [Google Scholar] [CrossRef]
- Wright, J.S.; Shadnia, H.; Chepelev, L.L. Stability of carbon-centered radicals: Effect of functional groups on the energetics of addition of molecular oxygen. J. Comput. Chem. 2009, 30, 1016–1026. [Google Scholar] [CrossRef]
- Gryn’Ova, G.; Hodgson, J.L.; Coote, M.L. Revising the mechanism of polymer autooxidation. Org. Biomol. Chem. 2010, 9, 480–490. [Google Scholar] [CrossRef]
- Greenhalgh, D.J.; Williams, A.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R. Adhesion of film coatings to tablet surfaces—A theoretical approach based on solubility parameters. Int. J. Pharm. 1988, 41, 219–222. [Google Scholar] [CrossRef]
- Rowe, R.C. Interactions in coloured powders and tablet formulations: A theoretical approach based on solubility parameters. Int. J. Pharm. 1989, 53, 47–51. [Google Scholar] [CrossRef]
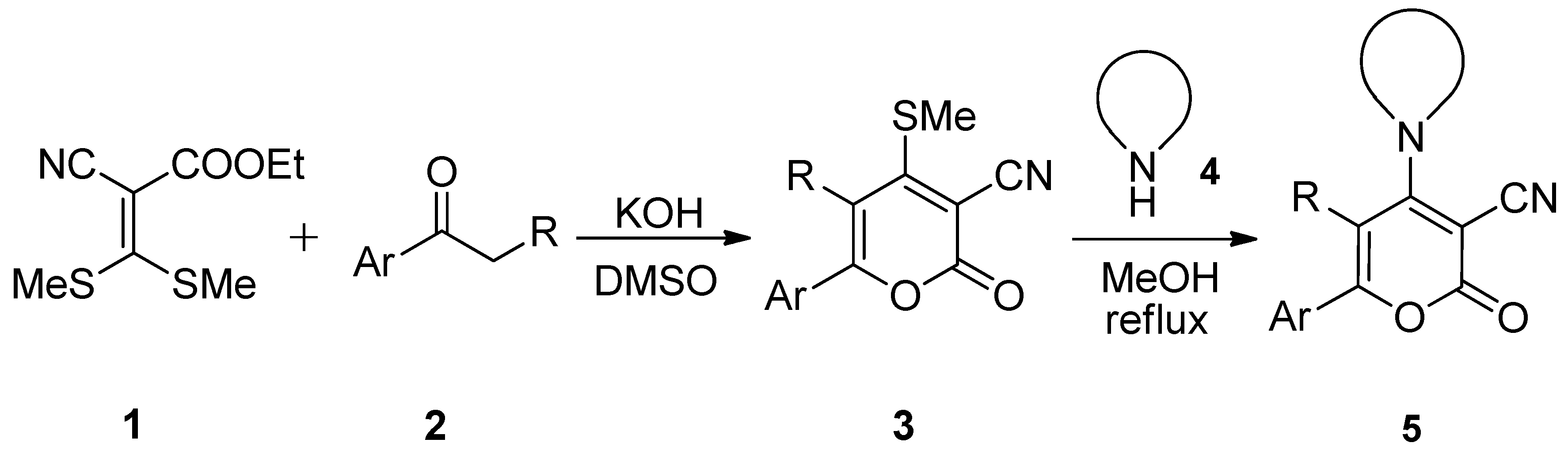


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure 7/11 | Time (min) | Yield (%) |
|---|---|---|---|
| RS-1 | 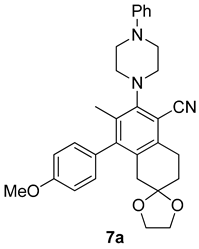 | 37 | 75 |
| RS-2 |  | 35 | 82 |
| RS-3 | 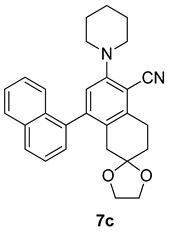 | 26 | 82 |
| RS-4 | 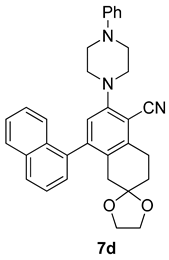 | 38 | 79 |
| RS-5 | 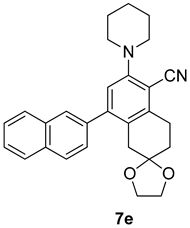 | 32 | 80 |
| RS-6 |  | 60 | 84 |
| Molecules | Interaction Energy [kcal/mol] | Average Number of Hydrogen Bonds |
|---|---|---|
| RS-1 | −65.175 | 3.020 |
| RS-2 | −63.523 | 3.024 |
| RS-3 | −75.854 | 3.019 |
| RS-4 | −76.809 | 4.057 |
| RS-5 | −70.066 | 3.127 |
| RS-6 | −66.056 | 2.925 |
| Molecules | [MPa1/2] |
|---|---|
| RS1 | 18.825 |
| RS2 | 18.663 |
| RS3 | 19.397 |
| RS4 | 19.264 |
| RS5 | 19.021 |
| RS6 | 19.513 |
| PVP | 18.515 |
| Maltose | 28.564 |
| Sorbitol | 32.425 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetgaonkar, S.E.; Kollur, S.P.; Pillai, R.R.; Thangavel, K.; Armaković, S.J.; Armaković, S.; Shivamallu, C.; Amachawadi, R.G.; Syed, A.; Elgorban, A.M.; et al. Investigation of Pharmaceutical Importance of 2H-Pyran-2-One Analogues via Computational Approaches. Symmetry 2021, 13, 1619. https://doi.org/10.3390/sym13091619
Shetgaonkar SE, Kollur SP, Pillai RR, Thangavel K, Armaković SJ, Armaković S, Shivamallu C, Amachawadi RG, Syed A, Elgorban AM, et al. Investigation of Pharmaceutical Importance of 2H-Pyran-2-One Analogues via Computational Approaches. Symmetry. 2021; 13(9):1619. https://doi.org/10.3390/sym13091619
Chicago/Turabian StyleShetgaonkar, Samata E., Shiva Prasad Kollur, Renjith Raveendran Pillai, Karthick Thangavel, Sanja J. Armaković, Stevan Armaković, Chandan Shivamallu, Raghavendra G. Amachawadi, Asad Syed, Abdallah M. Elgorban, and et al. 2021. "Investigation of Pharmaceutical Importance of 2H-Pyran-2-One Analogues via Computational Approaches" Symmetry 13, no. 9: 1619. https://doi.org/10.3390/sym13091619
APA StyleShetgaonkar, S. E., Kollur, S. P., Pillai, R. R., Thangavel, K., Armaković, S. J., Armaković, S., Shivamallu, C., Amachawadi, R. G., Syed, A., Elgorban, A. M., Bahkali, A. H., & Singh, F. V. (2021). Investigation of Pharmaceutical Importance of 2H-Pyran-2-One Analogues via Computational Approaches. Symmetry, 13(9), 1619. https://doi.org/10.3390/sym13091619