Abstract
Resorcinarenes decorated with sulfonate groups are anionic in nature and water soluble with a hydrophobic electron-rich interior cavity. These receptors are shown to bind zwitterionic aromatic mono-N-oxides and cationic di-N-oxide salts with varying spacer lengths. Titration data fit a 1:1 binding isotherm for the mono-N-oxides and 2:1 binding isotherm for the di-N-oxides. The first binding constants for the di-N-oxides (K1: 104 M−1) are higher compared to the neutral mono-N-oxide (K: 103 M−1) due to enhanced electrostatic attraction from a receptor with an electron-rich internal cavity and cationic and electron deficient N-oxides. The interaction parameter α reveals positive cooperativity for the di-N-oxide with a four-carbon spacer and negative cooperativity for the di-N-oxides that have spacers with more four carbons. This is attributed to shape complementarity between the host and the guest.
1. Introduction
Resorcinarenes are acid-catalyzed condensation products of resorcinol and appropriate aldehydes [1]. As supramolecular synthetic receptors, resorcinarenes have extensively been studied in host–guest chemistry for the development of functional (bio)materials and sensors [2,3], to name a few. The ability to functionalize the electron-rich upper-rim C2-position and lower-rim alkyl chains led researchers to follow complex supramolecular functions and processes both in aqueous and organic solvents. Incorporating water soluble functional groups into resorcinarene receptor design offers useful applications in drug delivery [4], chemical separation [5], nanoparticles [6], and as gelators [7]. The most notable feature of the receptor is the shallow, electron-rich hydrophobic cavity that binds to the neutral and cationic guest through C–H⋯π, cation⋯π and π⋯π interactions [8]. This versatility inspired our lab to fundamentally study host–guest complexes using the resorcinarene cavity to extrapolate a structure–property relationship based on guest affinities, conformation, size, and shape [9].
The incorporation of sodium sulfonate groups at C2-positions renders resorcinarenes anionic and water soluble with an amphiphilic character. Appealingly, the negatively charged sulfonate oxygens reinforce the cationic guests by ancillary C–H⋯OSO2 interactions, a feature that is not available in C2-H-resorcinarenes. The methylene groups between the aromatic ring and the sulfonate groups create an elongated cavity that further helps to improve the guest selectivity [9].
The host–guest chemistry of aromatic N-oxides has become widespread in supramolecular chemistry applications. This is credited to the versatility of N-oxides as precursors for synthesizing functionalized pyridine [10], as organocatalysts to access aromatic aldehydes [11], and as ligands in coordination chemistry [12]. We recently investigated the host–guest chemistry of resorcinarenes and substituted aromatic mono- and rigid di-N-oxides in organic media [13]. The conformational geometry of the endo-cavity guest inspired the use of the resorcinarenes as a protecting group on aromatic N-oxides to tune the coordination sphere of copper (II) [14]. We recognize the space to expand the fundamental use of functionalized resorcinarenes as suitable receptors for N-oxides in aqueous environments.
Herein, we present a host–guest study of sodiumsulfonatomethyleneresorcinarene (1) with three zwitterionic aromatic mono-N-oxides (2–4), and three cationic di-N-oxide salts with different spacer lengths (5–7) in water (Figure 1). Our motivation for this study stems from the fact that (i) the elongated cavity compared to basic resorcinarene renders a narrower cavity aperture that influences guest selectivity [9], and that (ii), while aromatic mono-N-oxides are zwitterionic, the di-N-oxides have two cationic binding sites in solution. In effect, we sought answers to the following questions: (a) How does the binding of one site on the di-N-oxide influence binding affinity to the second site? (b) How does the length of the aliphatic spacer chain influence the binding of the resorcinarene to both sites?
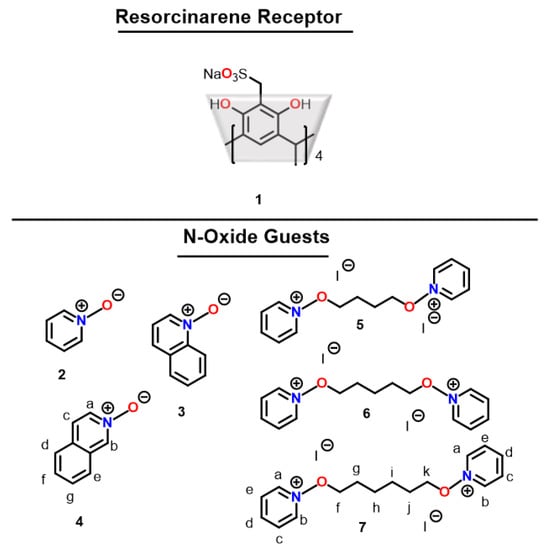
Figure 1.
Structure of the resorcinarene sulfonate receptor (1) and mono-N-oxides (2–4) and di-N-oxides (5–7).
2. Materials and Methods
N-oxides (2–4) and solvents used for syntheses, NMR experiments and Isothermal Titration Calorimetry (ITC) were purchased from Sigma Aldrich or Oakwood Chemicals (Estill SC, USA). The receptors (1) were synthesized according to reported procedures.1 All NMR and ITC experiments were carried out in H(D)2O or Tris buffer pH 7.4 at 298 K on a Bruker Avance 400 and Microcal VP-ITC 2000.
N-oxides 5–7 were synthesized according to reported procedures [15]. Acetonitrile solutions containing pyridine N-oxide (2.2 mmol) and corresponding diiodoalkanes (1.0 mmol) were stirred at reflux temperature overnight. The reaction mixture was cooled to room temperature. The salt precipitates formed were filtered and dried under high vacuum overnight.
Proton NMR Experiment: For sample preparation, stock solutions of the receptor 1 (2 mM), N-oxides (2 mM) were prepared in 100% D2O. For the pure 1–7 samples, 250 µL of the stock solution was transferred to an NMR tube and diluted with 250 µL of pure solvent to make a 1-mM sample concentration. For a 1:1 (1:2–7) mixture, 250 µL of 1 and 250 µL of 2–7 were pipetted into a clean NMR tube making 1:1 equimolar sample concentration of each component in the mixture.
The MicroCal VP-ITC instrument by Malvernwas used to determine the molar enthalpy (∆H) of complexation. Subsequent fitting of the data to a 1:1 and 1:2 binding model using Origin software provides an association constant (K), and a change in enthalpy (∆H) and entropy (∆S). The ITC experiment was carried out by filling the sample cell with guests 2–7 (0.25 mM), filling the syringe with host 1 (5.0 mM), and titrating via computer-automated injector at 298 K.
The equilibrium structures and properties of guest molecules were obtained from B3LYP-D3 calculations with a 6-31G** basis set within the implicit polarized continuum solvation model (PCM) water solvent.
3. Results and Discussion
3.1. NMR Spectroscopy
Guests 2–4 were purchased from commercial sources while guests 5–7 were synthesized by mixing 2:1 equivalent of pyridine N-oxide and the corresponding di-iodoalkane in acetonitrile [15]. 1H NMR spectroscopy was used for screening the affinity for the aromatic N-oxide guests (2–7) by the resorcinarene receptor (1). The up field shifts of the guest signals clearly indicate endo-complexation with the host. In solution, the complexes are in rapid equilibrium and there is a fast exchange process with the free components; therefore, only one set of signals are observed in the NMR spectrum. Nonetheless, by following the guest protons and comparing the degree of shielding, one can also predict which part of the guest is preferred in the hydrophobic cavity.
The symmetrical nature of the di-N-oxides means only one set of aromatic signals are seen in their NMR spectra. The aromatic proton signals on the di-N-oxides experience significant shielding compared to the methylene protons, suggesting aromatic C–H protons and resorcinarenes are stabilized via endo-cavity C–H⋯π interactions (Figure 2). In all complexes, the aromatic hydrogen para to the nitrogen atom is shielded the most, suggesting it resides deeper in the hydrophobic cavity. The aromatic para protons on di-N-oxides are considerably shielded (5@1, 1.46; 6@1, 0.93; 7@1, 1.22 Δppm) compared to the mono-N-oxide guests (2@1, 0.10; 3@1, 0.42; 4@1, 0.13 Δppm). Notably, the tight binding of di-N-oxides to the host cavity is due to the blockage of the N–O group by alkylation which hampers the O-atom from hydrogen bonding participation. Guests 3 and 4 aromatic protons experience higher shielding effects compared to 2, presumably due to the size complementarity of guests 3 and 4 (Figure 2). Furthermore, the movement of the aromatic proton of the resorcinarene host also highlights the cavity “breathing” that enables it to accommodate the N-oxides. Additionally, the reduction in the electron density due to bound guests shows up as a de-shielded effect in the NMR spectra. The cationic di-N-oxides cause a greater de-shielding of the host proton compared to the “neutral” mono-N-oxides.

Figure 2.
Section of NMR spectra of (a) pure host (1) (b) 1:1 Guest 4@1 (c) Pure guest 4 (d) 1:1 Guest 7@1 (e) Pure guest 7 in pure D2O.
3.2. Isothermal Titration Calorimetry (ITC)
The quantification of the binding affinity (K) and complexation-induced changes to thermodynamic parameters (ΔH, TΔS, and ΔG) were addressed using Isothermal Titration Calorimetry (ITC) in deionized water, and in a buffer system. Tris buffer (10 mM) at pH 7.4 was used as the physiological buffer system of choice and to eliminate the possibility of protonation-induced heat changes. For mono-N-oxide guests 3 and 4, all the ITC data were fitted to one set of sites binding model while ITC data for di-N-oxide guests 5–7 were fitted to two set of sequential binding sites model and the respective ITC titration traces are included in the Supporting Information. ITC data for guest 2 could not be fitted to any binding isotherm in water and buffer system without significant errors (Figures S7a and S8a). Notably, pyridine molecules are known to bind and sit in the hydrophobic cavity of the resorcinarene host 1 [16]. However, the mono-N-oxide analog 2 does not show such behavior due to a comparatively weaker binding. From the NMR data, mono-N-oxides 3 and 4 both sit in the hydrophobic cavity with the nitrogen atom skewed to face outwards from the cavity. In addition, their binding affinities and thermodynamic parameters of complexation in both water and buffer to be almost identical (Table 1). This suggest protonation is not an issue in the water system.

Table 1.
Thermodynamic binding parameters of formed complexes between the receptors and the guests by Isothermal Titration Calorimetry (ITC) in 10 mM Tris buffer, pH 7.4. a
ITC complexation data for di-N-oxide guests 5–7 reveal more about the binding of resorcinarene hosts to divalent di-N-oxides. The binding affinity between the receptor and mono-N-oxides are lower compared to the di-N-oxides. In solution, the di-N-oxides are positively charged compared to the zwitterionic mono-N-oxides thus benefiting from electrostatic attractions to the host in addition to C–H⋯π and π⋯π interactions. The –CH2– spacer length between the N-oxide (6 and 7) groups did not substantially change the strength of binding of the first or second host, both in the same order of magnitude among guests 6 and 7 (Table 1). However, for di-N-oxide 5, the first binding is similar to the mono-N-oxides but the second binding is one order of magnitude higher, contrary to di-N-oxides 6 and 7. The negative ΔH shows the complexation of guests 3–4 to be exothermic in buffer where all protonation issues have been negated. The system is largely enthalpy-driven, with small entropy compensations. Most importantly, binding energies show the host–guest complexations are all spontaneous at 298 K.
Cooperativity in binding represent change in site specific binding as a function of the reaction process [17]. In theory, the di-N-oxides aromatic cores should be able to bind to two different hosts 1 with near equal K1 and K2 values. However, in reality, all possible ligation states coexist in equilibrium in solution. The length of the –CH2– chain between two binding sites in a 2:1 complex affects the overall binding process (Table 1). The K1 values of 6@1, and 7@1 occur at an order of magnitude higher than the second binding site. However, for di-N-oxide 5 with a C4 spacer, the K2 value is of higher magnitude than the first (103 M−1 vs 104 M−1). The interaction parameter α quantifies the extent of cooperativity in the second binding site when the first one is already occupied in equilibrium. Cooperativity in binding can be determined from the interaction parameter α (α = 4K2/K1, where K1 and K2 are the first and second binding event, respectively) [18,19]. If α > 1, the system displays positive cooperativity, if α < 1, it displays negative cooperativity and if α = 1 it displays a non-cooperative process (Table 2) [19].
Table 2.
Complexation derived interaction parameter that describes cooperativity in binding constants.
The interaction parameter for 5@1 = 63, for 6@1 and 7@1 < 1 signifies positive cooperativity for binding di-N-oxide guests with a C4 spacer and a negative cooperativity for C5 and C6 spacers. This result shows that the C4 spacer fits perfectly inside the “capsular-like arrangement, namely 5@12. The C5 and C6 spacers induce conformational flexibility which negatively affects the cooperativity.
3.3. Computational Studies of the Di-N-Oxides
We performed computational modelling to get the equilibrium structures of the guest molecules from Density Functional Theory (DFT) B3LYP-D3 calculations with a 6-31G** basis set within the implicit polarized continuum solvation model (PCM) water solvent. In interactions of polar (and charged) species the cooperativity of interactions is dominated by the three-body polarization effect [20]. The three-body polarization effect can be both attractive and repulsive. The attractive (or cooperativity-enhancing) effects are in “head-to-tail” → → → dipole arrangements (can be linear, circular or bent). The repulsive three-body polarization occurs in → → ← situations (also linear, circular, or bent) because the counteracting effect of the field-induced moments on the central system by the terminal ones [21,22]. This effect causes negative cooperativity and is strongly dependent on the magnitude of the central dipole. If the central dipole moment vanishes, the three-body repulsion is effectively minimized. Our geometry optimizations of the guests 5, 6 and 7 show that the equilibrium di-N-oxide with a 4C spacer belongs to the symmetry group C2h. The molecules in this point group are nonpolar. The 5C spacer di-cation has C2v symmetry with a large dipole moment 4.38 D. With 6C spacer di-cation, there exists the possibility of a C2h structure, but this structure is not optimal, as our calculations show, and the bent C1 structure shown below (Table 3) is more stable by at least 0.4 kcal/mol. The C1 structure is also polar. So, the favorable cooperativity observed in ITC measurements is attributed to the symmetry of guest 5 which mandates the zero dipole moment and, in turn, reduces the three-body repulsive polarization effect (Table 3).
Table 3.
Properties of optimized guest molecules from DFT calculations. The geometry optimizations we performed in implicit polarized continuum solvation model (PCM) water solvent with the B3LYP-D3 functional and 6-31G** basis set.
4. Conclusions
The binding of aromatic mono-N-oxide and di-N-oxides in water by a C1-tetrasodiumsulfonatomethyleneresorcinarene is presented. 1H-NMR reveals the significant shielding of aromatic protons, which is greater for di-N-oxides compared to their mono-N-oxide counterparts when located inside the electron-rich cavity. The binding processes quantified through ITC experiments demonstrate a much higher binding affinity for the di-N-oxides than mono-N-oxides. The first binding event is one order of magnitude stronger than the second for di-N-oxides. Positive cooperativity was observed for the di-N-oxide with a four-carbon spacer, while negative cooperativity was observed for di-N-oxides with more than four-carbon spacer. Computation studies of the structures of the di-N-oxides show that the four-carbon spaced one belongs to a symmetry group that mandates the zero dipole moment, which, in turn, reduces the three-body repulsive polarization effect. This work shows that resorcinarenes possess a suitable binding pocket for aromatic N-oxides in aqueous media, which should help accelerate diagnostic applications of biological aromatic-N-oxides.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-8994/12/11/1751/s1, Figures S1–S7: 1H NMR spectra (D2O, 298 K) of host–guest complexes, Figures S7 and S8: ITC traces of host–guest complexes, Table S1: Thermodynamic binding parameters of formed complexes between the receptors and the guests in H2O by ITC., Table S2: Complexation derived interaction parameter (α) that describes cooperativity in binding constants for thermodynamics in deionized H2O, Table S3: Equilibrium structures and properties of guest molecules from B3LYP-D3 calculations with a 6-31G** basis set within the implicit PCM water solvent; the electrostatic potential scale shown is in kJ/mole.
Author Contributions
Conceptualization, N.K.B. and R.P.; methodology, K.T., N.S., B.E., J.F., L.Y., M.M.S.; software, M.M.S.; validation, K.T., N.S., B.E., J.F., L.Y. and M.M.S.; writing—original draft preparation, K.T., writing—review and editing, K.T., R.P., M.M.S., N.K.B.; supervision, N.K.B.; project administration, N.K.B.; funding acquisition, N.K.B., K.T., R.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Academy of Finland, grant number 298817.
Acknowledgments
The authors gratefully acknowledge the financial support from the Provost Graduate Student Research grant and Oakland University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Timmerman, P.; Verboom, W.; Reinhoudt, D.N. Resorcinarenes. Tetrahedron 1996, 52, 2663–2704. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Díez, I.; Taimoory, S.M.; Meister, D.; Feig, A.I.; Trant, J.F.; Ras, R.H.A.; Rissanen, K. High-Affinity and Selective Detection of Pyrophosphate in Water by a Resorcinarene Salt Receptor. Chem. Sci. 2018, 9, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Beyeh, N.K.; Nonappa; Liljeström, V.; Mikkilä, J.; Korpi, A.; Bochicchio, D.; Pavan, G.M.; Ikkala, O.; Ras, R.H.A.; Kostiainen, M.A. Crystalline Cyclophane-Protein Cage Frameworks. ACS Nano 2018, 12, 8029–8036. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Papachristou, A.; Ho, T.M.H.; Hine, J.; Curtis, A.D.M. Investigation into Drug Solubilisation Potential of Sulfonated Calix [4] Resorcinarenes. J. Nanomed. Nanotechnol. 2016, 7, 1000370. [Google Scholar] [CrossRef]
- Li, N.; Harrison, R.G.; Lamb, J.D. Application of Resorcinarene Derivatives in Chemical Separations. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 39–60. [Google Scholar] [CrossRef]
- Kim, B.; Balasubramanian, R.; Pérez-Segarra, W.; Wei, A.; Decker, B.; Mattay, J. Self-Assembly of Resorcinarene-Stabilized Gold Nanoparticles: Influence of the Macrocyclic Headgroup. Supramol. Chem. 2005, 17, 173–180. [Google Scholar] [CrossRef]
- Haines, S.R.; Harrison, R.G. Novel Resorcinarene-Based PH-Triggered Gelator. Chem. Commun. 2002, 2, 2846–2847. [Google Scholar] [CrossRef]
- Schneider, H.J.; Schneider, U. The Host-Guest Chemistry of Resorcinarenes. J. Incl. Phenom. Mol. Recognit. Chem. 1994, 19, 67–83. [Google Scholar] [CrossRef]
- Twum, K.; Rautiainen, J.M.; Yu, S.; Truong, K.N.; Feder, J.; Rissanen, K.; Puttreddy, R.; Beyeh, N.K. Host-Guest Interactions of Sodiumsulfonatomethyleneresorcinarene and Quaternary Ammonium Halides: An Experimental-Computational Analysis of the Guest Inclusion Properties. Cryst. Growth Des. 2020, 20, 2367–2376. [Google Scholar] [CrossRef]
- Andersson, H.; Almqvist, F.; Olsson, R. Synthesis of 2-Substituted Pyridines via a Regiospecific Alkylation, Alkynylation, and Arylation of Pyridine N-Oxides. Org. Lett. 2007, 9, 1335–1337. [Google Scholar] [CrossRef]
- Pignataro, L.; Benaglia, M.; Annunziata, R.; Cinquini, M.; Cozzi, F. Structurally Simple Pyridine N-Oxides as Efficient Organocatalysts for the Enantioselective Allylation of Aromatic Aldehydes. J. Org. Chem. 2006, 71, 1458–1463. [Google Scholar] [CrossRef]
- Bullock, S.J.; Harding, L.P.; Moore, M.P.; Mills, A.; Piela, S.A.F.; Rice, C.R.; Towns-Andrews, L.; Whitehead, M. Synthesis of Ligands Containing N-Oxide Donor Atoms and Their Assembly into Metallosupramolecular Structures. Dalton Trans. 2013, 42, 5805–5811. [Google Scholar] [CrossRef][Green Version]
- Puttreddy, R.; Beyeh, N.K.; Taimoory, S.M.; Meister, D.; Trant, J.F.; Rissanen, K. Host–Guest Complexes of Conformationally Flexible C -Hexyl-2-Bromoresorcinarene and Aromatic N-Oxides: Solid-State, Solution and Computational Studies. Beilstein J. Org. Chem. 2018, 14, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Beyeh, N.K.; Puttreddy, R. Methylresorcinarene: A Reaction Vessel to Control the Coordination Geometry of Copper(Ii) in Pyridine N-Oxide Copper(Ii) Complexes. Dalton Trans. 2015, 44, 9881–9886. [Google Scholar] [CrossRef] [PubMed]
- Feely, W.E.; Beavers, E.M. Cyanation of Amine Oxide Salts. A New Synthesis of Cyanopyridines. J. Am. Chem. Soc. 1959, 81, 4004–4007. [Google Scholar] [CrossRef]
- Taimoory, S.M.; Twum, K.; Dashti, M.; Pan, F.; Lahtinen, M.; Rissanen, K.; Puttreddy, R.; Trant, J.F.; Beyeh, N.K. Bringing a Molecular plus One: Synergistic Binding Creates Guest-Mediated Three-Component Complexes. J. Org. Chem. 2020, 85, 5884–5894. [Google Scholar] [CrossRef]
- Cattoni, D.I.; Chara, O.; Kaufman, S.B.; Flecha, F.L.G. Cooperativity in Binding Processes: New Insights from Phenomenological Modeling. PLoS ONE 2015, 10, e0146043. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P. Determining Association Constants from Titration Experiments in Supramolecular Chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- Hunter, C.A.; Anderson, H.L. What Is Cooperativity? Angew. Chem. Int. Ed. 2009, 48, 7488–7499. [Google Scholar] [CrossRef]
- Szczȩśniak, M.M.; Chałasiński, G. Ab Initio Calculations of Nonadditive Effects. J. Mol. Struct. THEOCHEM 1992, 261, 37–54. [Google Scholar] [CrossRef]
- Szczȩśniak, M.M.; Chałasiński, G.; Piecuch, P. The Nonadditive Interactions in the Ar2HF and Ar2HCl Clusters: An Ab Initio Study. J. Chem. Phys. 1993, 99, 6732–6741. [Google Scholar] [CrossRef]
- Hapka, M.; Rajchel, Ł.; Modrzejewski, M.; Schäffer, R.; Chałasiński, G.; Szczȩśniak, M.M. The Nature of Three-Body Interactions in DFT: Exchange and Polarization Effects. J. Chem. Phys. 2017, 147. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).