Differential Gene Expression with an Emphasis on Floral Organ Size Differences in Natural and Synthetic Polyploids of Nicotiana tabacum (Solanaceae)

 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. Flower Size and Cell Size
2.3. Transcriptome Sequencing and Analyses
3. Results
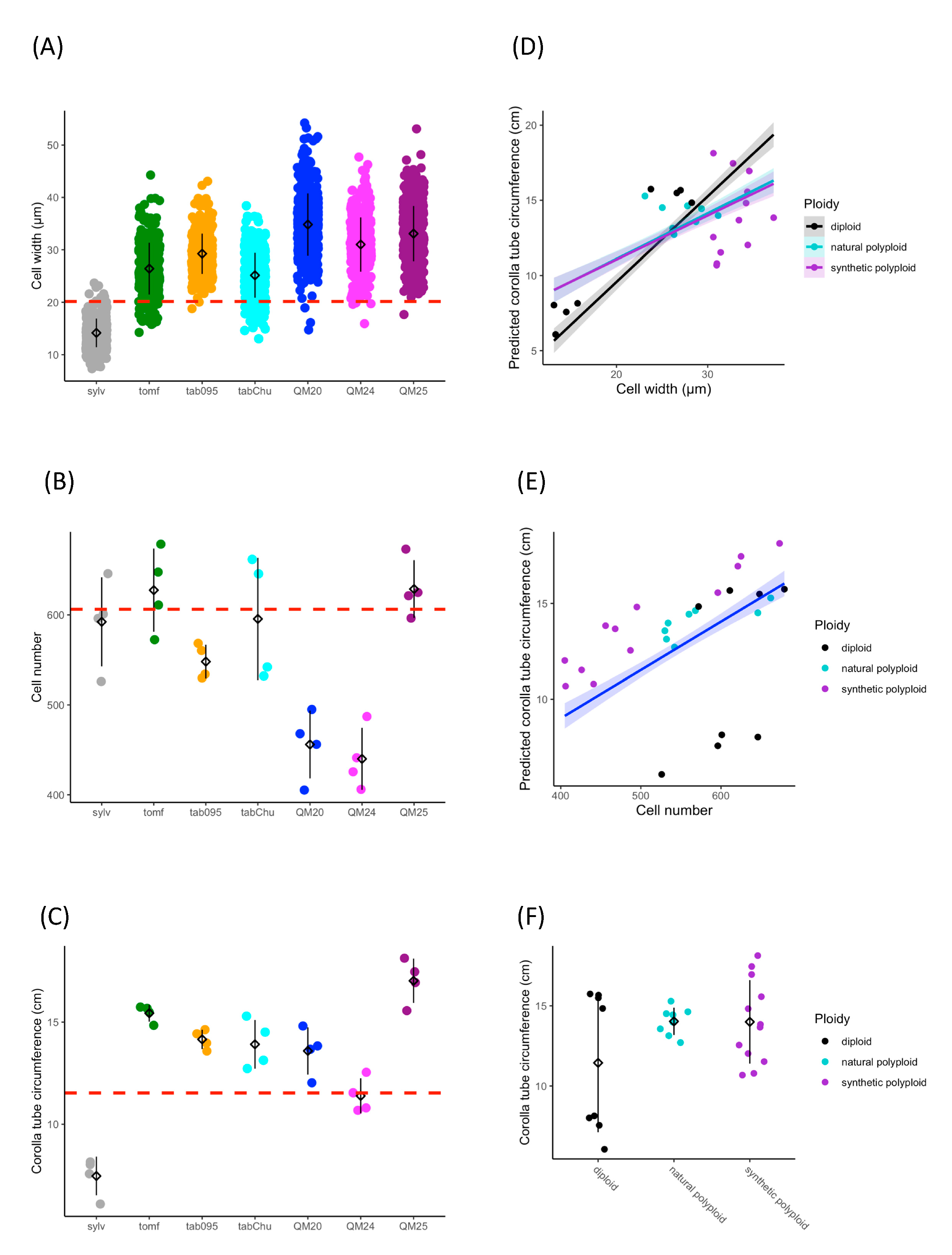
3.1. Cell width, Cell Number, and Ploidy Positively Influence Corolla Tube Circumference
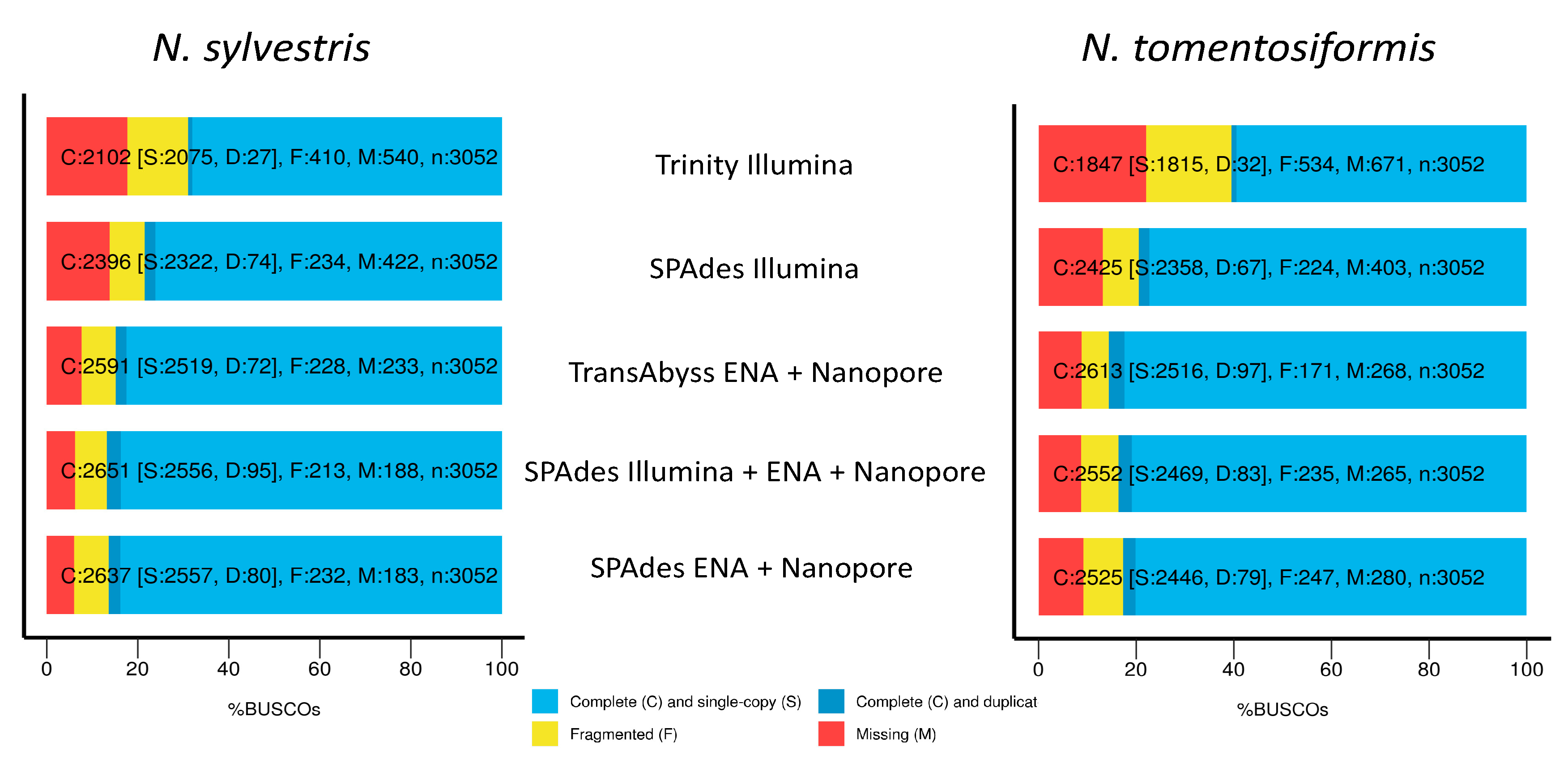
3.2. Transcriptome Assemblies
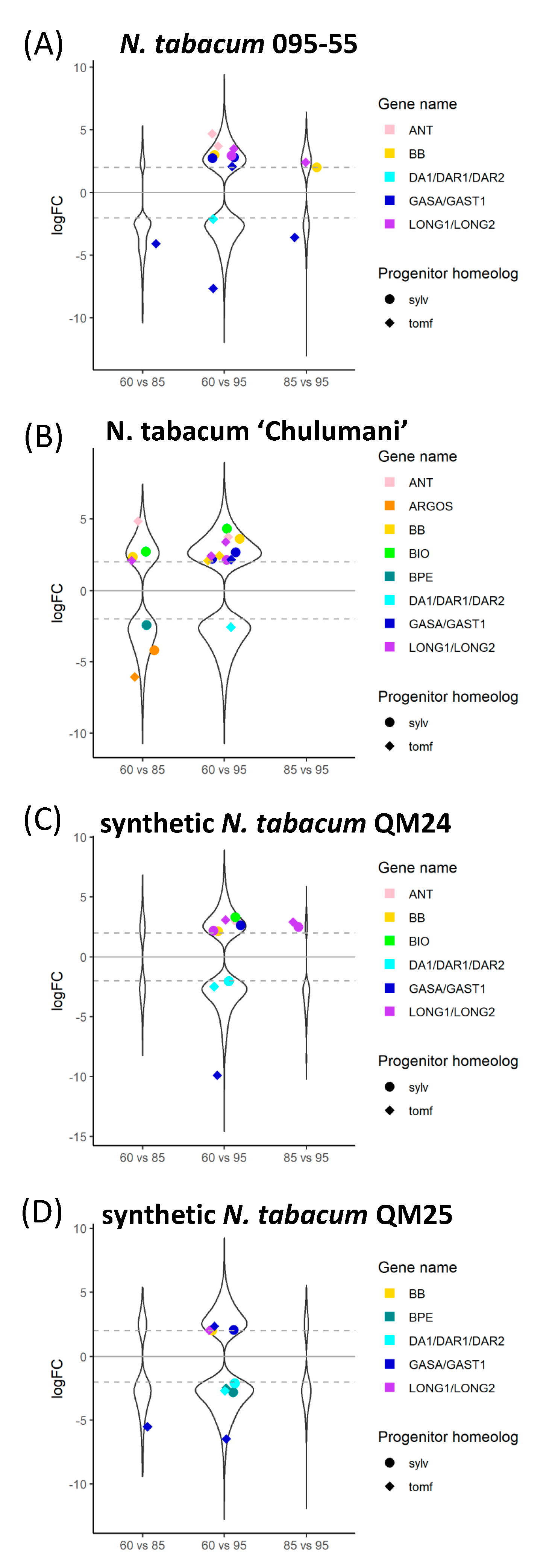
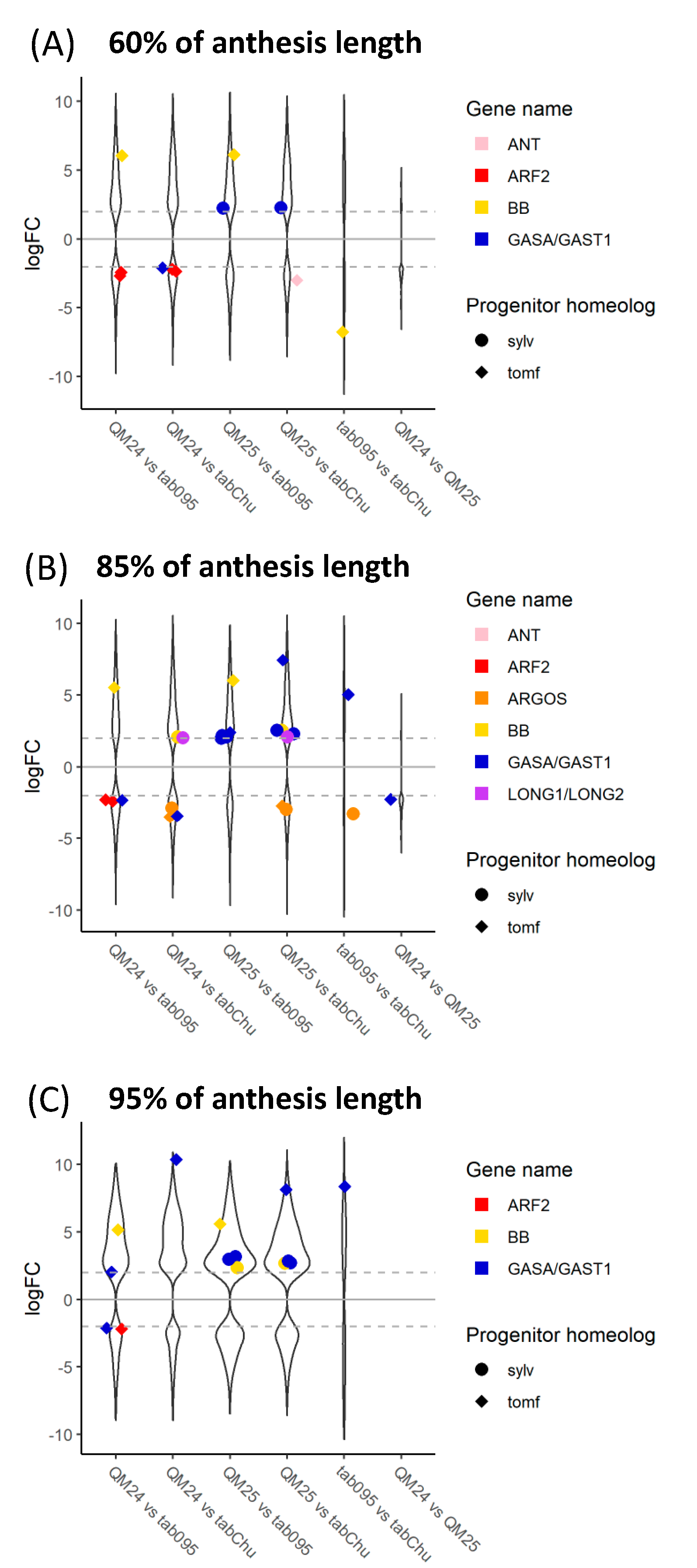
3.3. Differential Expression
4. Discussion
4.1. Differences in Expression
4.2. De Novo Transcriptome Assembly
4.3. Cell Size Increase Results in Wider Corolla Tubes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Mizukami, Y. A matter of size: Developmental control of organ size in plants. Curr. Opin. Plant Biol. 2001, 4, 533–539. [Google Scholar] [CrossRef]
- Breuninger, H.; Lenhard, M. Control of tissue and organ growth in plants. Curr. Top. Dev. Biol. 2010, 91, 185–220. [Google Scholar] [PubMed]
- Irish, V.F. The Arabidopsis petal: A model for plant organogenesis. Trends Plant Sci. 2008, 13, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, J.; Lenhard, M. Regulation of plant lateral-organ growth by modulating cell number and size. Curr. Opin. Plant Biol. 2014, 17, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Hagemann, W. The Relationship of Cell and Organism in Vascular Plants. Bioscience 1991, 41, 693–703. [Google Scholar] [CrossRef]
- Feng, G.; Qin, Z.; Yan, J.; Zhang, X.; Hu, Y. Arabidopsis organ size related1 regulates organ growth and final organ size in orchestration with ARGOS and ARL. New Phytol. 2011, 191, 635–646. [Google Scholar] [CrossRef]
- Varaud, E.; Brioudes, F.; Szécsi, J.; Leroux, J.; Brown, S.; Perrot-Rechenmann, C.; Bendahmane, M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 2011, 23, 973–983. [Google Scholar] [CrossRef]
- Xu, R.; Li, Y. Control of final organ size by Mediator complex subunit 25 in Arabidopsis thaliana. Development 2011, 138, 4545–4554. [Google Scholar] [CrossRef]
- Martin, C.; Gerats, T. Control of Pigment Biosynthesis Genes during Petal Development. Plant Cell 1993, 5, 1253–1264. [Google Scholar] [CrossRef]
- Stuurman, J.; Hoballah, M.E.; Broger, L.; Moore, J.; Basten, C.; Kuhlemeier, C. Dissection of floral pollination syndromes in Petunia. Genetics 2004, 168, 1585–1599. [Google Scholar] [CrossRef]
- Fenster, C.B.; Armbruster, W.S.; Wilson, P.; Dudash, M.R.; Thomson, J.D. Pollination Syndromes and Floral Specialization. Annu. Rev. Ecol. Evol. Syst. 2004, 35, 375–403. [Google Scholar] [CrossRef]
- Sargent, R.D.; Goodwillie, C.; Kalisz, S.; Ree, R.H. Phylogenetic evidence for a flower size and number trade-off. Am. J. Bot. 2007, 94, 2059–2062. [Google Scholar] [CrossRef] [PubMed]
- Goodwillie, C.; Sargent, R.D.; Eckert, C.G.; Elle, E.; Geber, M.A.; Johnston, M.O.; Kalisz, S.; Moeller, D.A.; Ree, R.H.; Vallejo-Marin, M.; et al. Correlated evolution of mating system and floral display traits in flowering plants and its implications for the distribution of mating system variation. New Phytol. 2010, 185, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Moyroud, E.; Glover, B.J. The Evolution of Diverse Floral Morphologies. Curr. Biol. 2017, 27, R941–R951. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, V.M. The effects of floral display on pollinator visitation vary among populations ofPhacelia linearis (Hydrophyllaceae). Evol. Ecol. 1991, 5, 370–384. [Google Scholar] [CrossRef]
- Kettle, C.J.; Maycock, C.R.; Ghazoul, J.; Hollingsworth, P.M.; Khoo, E.; Sukri, R.S.H.; Burslem, D.F.R.P. Ecological implications of a flower size/number trade-off in tropical forest trees. PLoS ONE 2011, 6, e16111. [Google Scholar] [CrossRef]
- Solís-Montero, L.; Vallejo-Marín, M. Does the morphological fit between flowers and pollinators affect pollen deposition? An experimental test in a buzz-pollinated species with anther dimorphism. Ecol. Evol. 2017, 7, 2706–2715. [Google Scholar] [CrossRef]
- Krizek, B.A.; Anderson, J.T. Control of flower size. J. Exp. Bot. 2013, 64, 1427–1437. [Google Scholar] [CrossRef]
- Stebbins, G.L. Flowering Plants: Evolution above the Species Level; Belknap Press: Cambridge, MA, USA, 1974. [Google Scholar]
- Glover, B.J. Understanding Flowers and Flowering: An Integrated Approach; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Ojeda, I.; Santos-Guerra, A.; Caujapé-Castells, J.; Jaén-Molina, R.; Marrero, Á.; Cronk, Q.C.B. Comparative Micromorphology of Petals in Macaronesian Lotus (Leguminosae) Reveals a Loss of Papillose Conical Cells during the Evolution of Bird Pollination. Int. J. Plant Sci. 2012, 173, 365–374. [Google Scholar] [CrossRef]
- Krizek, B.A. Making bigger plants: Key regulators of final organ size. Curr. Opin. Plant Biol. 2009, 12, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Gast, R.T.; Gopalraj, M.; Olszewski, N.E. Characterization of a shoot-specific, GA3- and ABA-regulated gene from tomato. Plant J. 1992, 2, 153–159. [Google Scholar] [PubMed]
- Herzog, M.; Dorne, A.M.; Grellet, F. GASA, a gibberellin-regulated gene family from Arabidopsis thaliana related to the tomato GAST1 gene. Plant Mol. Biol. 1995, 27, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Kotilainen, M.; Helariutta, Y.; Mehto, M.; Pollanen, E.; Albert, V.A.; Elomaa, P.; Teeri, T.H. GEG participates in the regulation of cell and organ shape during corolla and carpel development in gerbera hybrida. Plant Cell 1999, 11, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Disch, S.; Anastasiou, E.; Sharma, V.K.; Laux, T.; Fletcher, J.C.; Lenhard, M. The E3 Ubiquitin Ligase BIG BROTHER Controls Arabidopsis Organ Size in a Dosage-Dependent Manner. Curr. Biol. 2006, 16, 272–279. [Google Scholar] [CrossRef]
- Szécsi, J.; Joly, C.; Bordji, K.; Varaud, E.; Cock, J.M.; Dumas, C.; Bendahmane, M. BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 2006, 25, 3912–3920. [Google Scholar] [CrossRef]
- Lee, Y.K.; Kim, G.-T.; Kim, I.-J.; Park, J.; Kwak, S.-S.; Choi, G.; Chung, W.-I. LONGIFOLIA1 and LONGIFOLIA2, two homologous genes, regulate longitudinal cell elongation in Arabidopsis. Development 2006, 133, 4305–4314. [Google Scholar] [CrossRef]
- Kim, G.-T.; Shoda, K.; Tsuge, T.; Cho, K.-H.; Uchimiya, H.; Yokoyama, R.; Nishitani, K.; Tsukaya, H. The ANGUSTIFOLIA gene of Arabidopsis, a plant CtBP gene, regulates leaf-cell expansion, the arrangement of cortical microtubules in leaf cells and expression of a gene involved in cell-wall formation. EMBO J. 2002, 21, 1267–1279. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, Q.; Chua, N.-H. The Arabidopsis auxin-inducible gene ARGOS controls lateral organ size. Plant Cell 2003, 15, 1951–1961. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Wei, J.; Winkler, C.; Goncalves-Butruille, M.; Weers, B.P.; Cerwick, S.F.; Dieter, J.A.; Duncan, K.E.; Howard, R.J.; et al. Maize ARGOS1 (ZAR1) transgenic alleles increase hybrid maize yield. J. Exp. Bot. 2014, 65, 249–260. [Google Scholar] [CrossRef]
- Krizek, B. AINTEGUMENTA and AINTEGUMENTA-LIKE6 act redundantly to regulate Arabidopsis floral growth and patterning. Plant Physiol. 2009, 150, 1916–1929. [Google Scholar] [CrossRef]
- Krizek, B.A.; Eaddy, M. AINTEGUMENTA-LIKE6 regulates cellular differentiation in flowers. Plant Mol. Biol. 2012, 78, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.M.; Nagpal, P.; Young, J.C.; Hagen, G.; Guilfoyle, T.J.; Reed, J.W. AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development 2005, 132, 4563–4574. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, E.; Kenz, S.; Gerstung, M.; MacLean, D.; Timmer, J.; Fleck, C.; Lenhard, M. Control of plant organ size by KLUH/CYP78A5-dependent intercellular signaling. Dev. Cell 2007, 13, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Horváth, B.M.; Magyar, Z.; Zhang, Y.; Hamburger, A.W.; Bakó, L.; Visser, R.G.F.; Bachem, C.W.B.; Bögre, L. EBP1 regulates organ size through cell growth and proliferation in plants. EMBO J. 2006, 25, 4909–4920. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, L.; Corke, F.; Smith, C.; Bevan, M.W. Control of final seed and organ size by the DA1 gene family in Arabidopsis thaliana. Genes Dev. 2008, 22, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, L.; Lu, Y.; Wu, Y.; Dumenil, J.; Zhu, Z.; Bevan, M.W.; Li, Y. The ubiquitin receptors DA1, DAR1, and DAR2 redundantly regulate endoreduplication by modulating the stability of TCP14/15 in Arabidopsis. Plant Cell 2015, 27, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Landoni, M.; Cassani, E.; Pilu, R. Arabidopsis thaliana plants overexpressing Ramosa1 maize gene show an increase in organ size due to cell expansion. Sex. Plant Reprod. 2007, 20, 191–198. [Google Scholar] [CrossRef]
- Li, X.; Liu, W.; Zhuang, L.; Zhu, Y.; Wang, F.; Chen, T.; Yang, J.; Ambrose, M.; Hu, Z.; Weller, J.L.; et al. BIGGER ORGANS and ELEPHANT EAR-LIKE LEAF1 control organ size and floral organ internal asymmetry in pea. J. Exp. Bot. 2019, 70, 179–191. [Google Scholar] [CrossRef]
- Roberts, W.R.; Roalson, E.H. Comparative transcriptome analyses of flower development in four species of Achimenes (Gesneriaceae). BMC Genom. 2017, 18, 240. [Google Scholar] [CrossRef]
- Almeida, A.M.R.; Piñeyro-Nelson, A.; Yockteng, R.B.; Specht, C.D. Comparative analysis of whole flower transcriptomes in the Zingiberales. PeerJ 2018, 6, e5490. [Google Scholar] [CrossRef]
- He, W.; Chen, Y.; Gao, M.; Zhao, Y.; Xu, Z.; Cao, P.; Zhang, Q.; Jiao, Y.; Li, H.; Wu, L.; et al. Transcriptome Analysis of Litsea cubeba Floral Buds Reveals the Role of Hormones and Transcription Factors in the Differentiation Process. G3 Genes Genomes Genet. 2018, 8, 1103–1114. [Google Scholar]
- Wang, S.; Li, Z.; Jin, W.; Fang, Y.; Yang, Q.; Xiang, J. Transcriptome analysis and identification of genes associated with flower development in Rhododendron pulchrum Sweet (Ericaceae). Gene 2018, 679, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Zhu, C.; Zhou, Y.; Niu, X.; Miao, M.; Tang, X.; Chen, F.; Zhao, W.; Liu, Y. Transcriptome analysis of differentially expressed unigenes involved in flavonoid biosynthesis during flower development of Chrysanthemum morifolium “Chuju”. Sci. Rep. 2018, 8, 13414. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Ding, L.; Song, A.; Shen, F.; Jiang, J.; Chen, S.; Chen, F. Transcriptomic and hormone analyses reveal mechanisms underlying petal elongation in Chrysanthemum morifolium “Jinba”. Plant Mol. Biol. 2017, 93, 593–606. [Google Scholar] [CrossRef]
- Cohen, J.I. De novo Sequencing and Comparative Transcriptomics of Floral Development of the Distylous Species Lithospermum multiflorum. Front. Plant Sci. 2016, 7, 7. [Google Scholar] [CrossRef]
- Liqin, G.; Jianguo, Z.; Xiaoxia, L.; Guodong, R. Polyploidy-related differential gene expression between diploid and synthesized allotriploid and allotetraploid hybrids of Populus. Mol. Breed. 2019, 39, 69. [Google Scholar] [CrossRef]
- Landis, J.B.; Soltis, D.E.; Soltis, P.S. Comparative transcriptomic analysis of the evolution and development of flower size in Saltugilia (Polemoniaceae). BMC Genom. 2017, 18, 475. [Google Scholar] [CrossRef]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar]
- Landis, J.B.; Soltis, D.E.; Li, Z.; Marx, H.E.; Barker, M.S.; Tank, D.C.; Soltis, P.S. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 2018, 105, 348–363. [Google Scholar] [CrossRef]
- Schneider, H.; Liu, H.-M.; Chang, Y.-F.; Ohlsen, D.; Perrie, L.R.; Shepherd, L.; Kessler, M.; Karger, D.N.; Hennequin, S.; Marquardt, J.; et al. Neo- and Paleopolyploidy contribute to the species diversity of Asplenium the most species-rich genus of ferns: Polyploidy in Asplenium. J. Syst. Evol. 2017, 55, 353–364. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, F.-G.; Zhang, J.; Shang, H.; Liu, L.; Wang, H.; Zhao, G.-H.; Shen, H.; Yan, Y.-H. Dating Whole Genome Duplication in Ceratopteris thalictroides and Potential Adaptive Values of Retained Gene Duplicates. Int. J. Mol. Sci. 2019, 20, 1926. [Google Scholar] [CrossRef] [PubMed]
- Han, T.-S.; Zheng, Q.-J.; Onstein, R.E.; Rojas-Andrés, B.M.; Hauenschild, F.; Muellner-Riehl, A.N.; Xing, Y.-W. Polyploidy promotes species diversification of Allium through ecological shifts. New Phytol. 2019, 225, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.A. Plant speciation in the age of climate change. Ann. Bot. 2019, 124, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.D. The duration of meiosis. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1971, 178, 277–299. [Google Scholar]
- Beaulieu, J.M.; Leitch, I.J.; Patel, S.; Pendharkar, A.; Knight, C.A. Genome size is a strong predictor of cell size and stomatal density in angiosperms. New Phytol. 2008, 179, 975–986. [Google Scholar] [CrossRef]
- Snodgrass, S.J.; Jareczek, J.; Wendel, J.F. An examination of nucleotypic effects in diploid and polyploid cotton. AoB Plants 2017, 9, 9. [Google Scholar] [CrossRef][Green Version]
- Doyle, J.J.; Coate, J.E. Polyploidy, the Nucleotype, and Novelty: The Impact of Genome Doubling on the Biology of the Cell. Int. J. Plant Sci. 2019, 180, 1–52. [Google Scholar] [CrossRef]
- Roddy, A.B.; Théroux-Rancourt, G.; Abbo, T.; Benedetti, J.W.; Brodersen, C.R.; Castro, M.; Castro, S.; Gilbride, A.B.; Jensen, B.; Jiang, G.-F.; et al. The Scaling of Genome Size and Cell Size Limits Maximum Rates of Photosynthesis with Implications for Ecological Strategies. Int. J. Plant Sci. 2020, 181, 75–87. [Google Scholar] [CrossRef]
- Katagiri, Y.; Hasegawa, J.; Fujikura, U.; Hoshino, R.; Matsunaga, S.; Tsukaya, H. The coordination of ploidy and cell size differs between cell layers in leaves. Development 2016, 143, 1120–1125. [Google Scholar] [CrossRef]
- Coate, J.E.; Luciano, A.K.; Seralathan, V.; Minchew, K.J.; Owens, T.G.; Doyle, J.J. Anatomical, biochemical, and photosynthetic responses to recent allopolyploidy in Glycine dolichocarpa (Fabaceae). Am. J. Bot. 2012, 99, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K. Stomatal Characteristics at Different Ploidy Levels in Coffea L. Ann. Bot. 1997, 80, 689–692. [Google Scholar] [CrossRef]
- Wong, C.; Murray, B.G. Variable changes in genome size associated with different polyploid events in Plantago (Plantaginaceae). J. Hered. 2012, 103, 711–719. [Google Scholar] [CrossRef][Green Version]
- Kondorosi, E.; Roudier, F.; Gendreau, E. Plant cell-size control: Growing by ploidy? Curr. Opin. Plant Biol. 2000, 3, 488–492. [Google Scholar] [CrossRef]
- Kudo, N.; Kimura, Y. Nuclear DNA endoreduplication during petal development in cabbage: Relationship between ploidy levels and cell size. J. Exp. Bot. 2002, 53, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto-Shirasu, K.; Roberts, K. “Big it up”: Endoreduplication and cell-size control in plants. Curr. Opin. Plant Biol. 2003, 6, 544–553. [Google Scholar] [CrossRef]
- Cheniclet, C.; Rong, W.Y.; Causse, M.; Frangne, N.; Bolling, L.; Carde, J.-P.; Renaudin, J.-P. Cell Expansion and Endoreduplication Show a Large Genetic Variability in Pericarp and Contribute Strongly to Tomato Fruit Growth. Plant Physiol. 2005, 139, 1984–1994. [Google Scholar] [CrossRef]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef]
- Simonin, K.A.; Roddy, A.B. Genome downsizing, physiological novelty, and the global dominance of flowering plants. PLoS Biol. 2018, 16, e2003706. [Google Scholar] [CrossRef]
- Brochmann, C.; Brysting, A.K.; Alsos, I.G.; Borgen, L.; Grundt, H.H.; Scheen, A.-C.; Elven, R. Polyploidy in arctic plants. Biol. J. Linn. Soc. Lond. 2004, 82, 521–536. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [PubMed]
- Te Beest, M.; Le Roux, J.J.; Richardson, D.M.; Brysting, A.K.; Suda, J.; Kubesová, M.; Pysek, P. The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. 2012, 109, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; Heidel-Fischer, H.M.; Bekaert, M.; Rota, J.; Glöckner, G.; Platts, A.E.; Heckel, D.G.; Der, J.P.; Wafula, E.K.; Tang, M.; et al. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA 2015, 112, 8362–8366. [Google Scholar] [CrossRef] [PubMed]
- Visger, C.J.; Germain-Aubrey, C.C.; Patel, M.; Sessa, E.B.; Soltis, P.S.; Soltis, D.E. Niche divergence between diploid and autotetraploid Tolmiea. Am. J. Bot. 2016, 103, 1396–1406. [Google Scholar] [CrossRef]
- Tate, J.A.; Symonds, V.V.; Doust, A.N.; Buggs, R.J.A.; Mavrodiev, E.; Majure, L.C.; Soltis, P.S.; Soltis, D.E. Synthetic polyploids of Tragopogon miscellus and T. mirus (Asteraceae): 60 Years after Ownbey’s discovery. Am. J. Bot. 2009, 96, 979–988. [Google Scholar] [CrossRef]
- Malek, M.A.; Ismail, M.R.; Rafii, M.Y.; Rahman, M. Synthetic Brassica napus L.: Development and studies on morphological characters, yield attributes, and yield. Sci. World J. 2012, 2012, 416901. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, T.; Li, X.; Duan, M.; Wang, J.; Qiu, Y.; Wang, H.; Song, J.; Shen, D. Interspecific hybridization, polyploidization, and backcross of Brassica oleracea var. alboglabra with B. rapa var. purpurea morphologically recapitulate the evolution of Brassica vegetables. Sci. Rep. 2016, 6, 18618. [Google Scholar] [CrossRef]
- Pavlíková, Z.; Paštová, L.; Münzbergová, Z. Synthetic polyploids in Vicia cracca: Methodology, effects on plant performance and aneuploidy. Plant Syst. Evol. 2017, 303, 827–839. [Google Scholar] [CrossRef]
- Wei, N.; Du, Z.; Liston, A.; Ashman, T.-L. Genome duplication effects on functional traits and fitness are genetic context and species dependent: Studies of synthetic polyploid Fragaria. Am. J. Bot. 2019. [Google Scholar] [CrossRef]
- Forrester, N.J.; Ashman, T.-L. Autopolyploidy alters nodule-level interactions in the legume-rhizobium mutualism. Am. J. Bot. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Knapp, S.; Cox, A.V.; Clarkson, J.J.; Butsko, Y.; Joseph, J.; Savolainen, V.; Parokonny, A.S. Molecular systematics, GISH and the origin of hybrid taxa in Nicotiana (Solanaceae). Ann. Bot. 2003, 92, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.J.; Knapp, S.; Garcia, V.F.; Olmstead, R.G.; Leitch, A.R.; Chase, M.W. Phylogenetic relationships in Nicotiana (Solanaceae) inferred from multiple plastid DNA regions. Mol. Phylogenet. Evol. 2004, 33, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.J.; Kelly, L.J.; Leitch, A.R.; Knapp, S.; Chase, M.W. Nuclear glutamine synthetase evolution in Nicotiana: Phylogenetics and the origins of allotetraploid and homoploid (diploid) hybrids. Mol. Phylogenet. Evol. 2010, 55, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.J.; Dodsworth, S.; Chase, M.W. Time-calibrated phylogenetic trees establish a lag between polyploidisation and diversification in Nicotiana (Solanaceae). Plant Syst. Evol. 2017, 303, 1–12. [Google Scholar] [CrossRef]
- Kelly, L.J.; Leitch, A.R.; Clarkson, J.J.; Hunter, R.B.; Knapp, S.; Chase, M.W. Intragenic recombination events and evidence for hybrid speciation in Nicotiana (Solanaceae). Mol. Biol. Evol. 2010, 27, 781–799. [Google Scholar] [CrossRef]
- Kelly, L.J.; Leitch, A.R.; Clarkson, J.J.; Knapp, S.; Chase, M.W. Reconstructing the complex evolutionary origin of wild allopolyploid tobaccos (Nicotiana section suaveolentes). Evolution 2013, 67, 80–94. [Google Scholar] [CrossRef]
- McCarthy, E.W.; Landis, J.B.; Kurti, A.; Lawhorn, A.J.; Chase, M.W.; Knapp, S.; Le Comber, S.C.; Leitch, A.R.; Litt, A. Early consequences of allopolyploidy alter floral evolution in Nicotiana (Solanaceae). BMC Plant Biol. 2019, 19, 162. [Google Scholar] [CrossRef]
- McCarthy, E.W.; Chase, M.W.; Knapp, S.; Litt, A.; Leitch, A.R.; Le Comber, S.C. Transgressive phenotypes and generalist pollination in the floral evolution of Nicotiana polyploids. Nat. Plants 2016, 2, 16119. [Google Scholar] [CrossRef]
- Zenil-Ferguson, R.; Burleigh, J.G.; Freyman, W.A.; Igić, B.; Mayrose, I.; Goldberg, E.E. Interaction among ploidy, breeding system and lineage diversification. New Phytol. 2019, 224, 1252–1265. [Google Scholar] [CrossRef]
- Fregonezi, J.N.; Turchetto, C.; Bonatto, S.L.; Freitas, L.B. Biogeographical history and diversification of Petunia and Calibrachoa (Solanaceae) in the Neotropical Pampas grassland. Bot. J. Linn. Soc. 2013, 171, 140–153. [Google Scholar] [CrossRef]
- Reck-Kortmann, M.; Silva-Arias, G.A.; Segatto, A.L.A.; Mäder, G.; Bonatto, S.L.; de Freitas, L.B. Multilocus phylogeny reconstruction: New insights into the evolutionary history of the genus Petunia. Mol. Phylogenet. Evol. 2014, 81, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Turchetto, C.; Maestri, R.; Freitas, L.B. Morphological characterization of sympatric and allopatric populations of Petunia axillaris and P. exserta (Solanaceae). Bot. J. Linn. Soc. 2020, 192, 550–567. [Google Scholar] [CrossRef]
- Edwards, E.J.; Chatelet, D.S.; Chen, B.-C.; Ong, J.Y.; Tagane, S.; Kanemitsu, H.; Tagawa, K.; Teramoto, K.; Park, B.; Chung, K.-F.; et al. Convergence, Consilience, and the Evolution of Temperate Deciduous Forests. Am. Nat. 2017, 190, S87–S104. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.J.; Edwards, E.J. Model clades are vital for comparative biology, and ascertainment bias is not a problem in practice: A response to Beaulieu and O’Meara (2018). Am. J. Bot. 2019, 106, 327–330. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- R Core team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Kuznetsova, A.; Brockhoff, P.; Christensen, R. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. Artic. 2017, 82, 1–26. [Google Scholar] [CrossRef]
- Lenth, R. Least-Squares Means: The R Package lsmeans. J. Stat. Softw. Artic. 2016, 69, 1–33. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.-R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Wang, J.R.; Holt, J.; McMillan, L.; Jones, C.D. FMLRC: Hybrid long read error correction using an FM-index. BMC Bioinform. 2018, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.; McMillan, L. Merging of multi-string BWTs with applications. Bioinformatics 2014, 30, 3524–3531. [Google Scholar] [CrossRef]
- Zhang, H.; Jain, C.; Aluru, S. A comprehensive evaluation of long read error correction methods. bioRxiv 2019, 519330. [Google Scholar] [CrossRef]
- Fu, S.; Wang, A.; Au, K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Sierro, N.; Battey, J.N.D.; Ouadi, S.; Bovet, L.; Goepfert, S.; Bakaher, N.; Peitsch, M.C.; Ivanov, N.V. Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 2013, 14, R60. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.D.; Fernandez-Pozo, N.; Drake-Stowe, K.; Humphry, M.; Evans, A.D.; Bombarely, A.; Allen, F.; Hurst, R.; White, B.; Kernodle, S.P.; et al. A reference genome for Nicotiana tabacum enables map-based cloning of homeologous loci implicated in nitrogen utilization efficiency. BMC Genom. 2017, 18, 448. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini-metagenomes from Highly Chimeric Reads. In Proceedings of the Research in Computational Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 158–170. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Polychronopoulos, D.; Lenhard, B. CNEr: A toolkit for exploring extreme noncoding conservation. PLoS Comput. Biol. 2019, 15, e1006940. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Landis, J.B.; O’Toole, R.D.; Ventura, K.L.; Gitzendanner, M.A.; Oppenheimer, D.G.; Soltis, D.E.; Soltis, P.S. The phenotypic and genetic underpinnings of flower size in Polemoniaceae. Front. Plant Sci. 2016, 6, 1144. [Google Scholar] [CrossRef]
- Zhurinsky, J.; Leonhard, K.; Watt, S.; Marguerat, S.; Bähler, J.; Nurse, P. A coordinated global control over cellular transcription. Curr. Biol. 2010, 20, 2010–2015. [Google Scholar] [CrossRef]
- Marguerat, S.; Bähler, J. Coordinating genome expression with cell size. Trends Genet. 2012, 28, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Ietswaart, R.; Rosa, S.; Wu, Z.; Dean, C.; Howard, M. Cell-Size-Dependent Transcription of FLC and Its Antisense Long Non-coding RNA COOLAIR Explain Cell-to-Cell Expression Variation. Cell Syst 2017, 4, 622–635.e9. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Rolfe, P.A.; Gifford, D.K.; Fink, G.R. Control of transcription by cell size. PLoS Biol. 2010, 8, e1000523. [Google Scholar] [CrossRef] [PubMed]
- Padovan-Merhar, O.; Nair, G.P.; Biaesch, A.G.; Mayer, A.; Scarfone, S.; Foley, S.W.; Wu, A.R.; Churchman, L.S.; Singh, A.; Raj, A. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol. Cell 2015, 58, 339–352. [Google Scholar] [CrossRef]
- Schoenfelder, K.P.; Fox, D.T. The expanding implications of polyploidy. J. Cell Biol. 2015, 209, 485–491. [Google Scholar] [CrossRef]
- Jaakola, L.; Määttä, K.; Pirttilä, A.M.; Törrönen, R.; Kärenlampi, S.; Hohtola, A. Expression of genes involved in anthocyanin biosynthesis in relation to anthocyanin, proanthocyanidin, and flavonol levels during bilberry fruit development. Plant Physiol. 2002, 130, 729–739. [Google Scholar] [CrossRef]
- Wilhelmsson, P.K.I.; Chandler, J.O.; Fernandez-Pozo, N.; Graeber, K.; Ullrich, K.K.; Arshad, W.; Khan, S.; Hofberger, J.A.; Buchta, K.; Edger, P.P.; et al. Usability of reference-free transcriptome assemblies for detection of differential expression: A case study on Aethionema arabicum dimorphic seeds. BMC Genom. 2019, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Holding, M.L.; Margres, M.J.; Mason, A.J.; Parkinson, C.L.; Rokyta, D.R. Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics. Toxins 2018, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-H.; Oyang, Y.-J.; Chen, C.-Y. Effect of de novo transcriptome assembly on transcript quantification. Sci. Rep. 2019, 9, 8304. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.W.; Landis, J.B.; Kurti, A.; Lawhorn, A.J.; Litt, A. The Genetic Basis of Flower Color Differences in Nicotiana tabacum. In The Tobacco Plant Genome; Ivanov, N.V., Sierro, N., Peitsch, M.C., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 175–193. ISBN 9783030294939. [Google Scholar]
- Francis, D.; Davies, M.S.; Barlow, P.W. A strong nucleotypic effect on the cell cycle regardless of ploidy level. Ann. Bot. 2008, 101, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.O.; Coate, J.E.; Singh, A.; Hong, L.; Bush, M.; Doyle, J.J.; Roeder, A.H.K. Ploidy and Size at Multiple Scales in the Arabidopsis Sepal. Plant Cell 2018, 30, 2308–2329. [Google Scholar] [CrossRef]
- Miller, M.; Zhang, C.; Chen, Z.J. Ploidy and Hybridity Effects on Growth Vigor and Gene Expression in Arabidopsis thaliana Hybrids and Their Parents. G3 Genes Genomes Genet. 2012, 2, 505–513. [Google Scholar] [CrossRef]
- Shao, J.; Chen, C.; Deng, X. In vitro induction of tetraploid in pomegranate (Punica granatum). Plant Cell Tissue Organ Cult. 2003, 75, 241–246. [Google Scholar] [CrossRef]
- Zhang, Z.; Dai, H.; Xiao, M.; Liu, X. In vitro induction of tetraploids in Phlox subulata L. Euphytica 2008, 159, 59–65. [Google Scholar] [CrossRef]
- Gantait, S.; Mandal, N.; Bhattacharyya, S.; Das, P.K. Induction and identification of tetraploids using in vitro colchicine treatment of Gerbera jamesonii Bolus cv. Sciella. Plant Cell Tissue Organ Cult. 2011, 106, 485. [Google Scholar] [CrossRef]
- Vamosi, J.C.; Goring, S.J.; Kennedy, B.F.; Mayberry, R.J.; Moray, C.M.; Neame, L.A.; Tunbridge, N.D.; Elle, E. Pollination, floral display, and the ecological correlates of polyploidy. Funct. Ecosyst. Communities 2007, 1, 1–9. [Google Scholar]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic Changes in Resynthesized Brassica napus and Their Effect on Gene Expression and Phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef] [PubMed]
- Leitch, I.J.; Hanson, L.; Lim, K.Y.; Kovarik, A.; Chase, M.W.; Clarkson, J.J.; Leitch, A.R. The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann. Bot. 2008, 101, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Münzbergová, Z. Colchicine application significantly affects plant performance in the second generation of synthetic polyploids and its effects vary between populations. Ann. Bot. 2017, 120, 329–339. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.W.; Arnold, S.E.J.; Chittka, L.; Le Comber, S.C.; Verity, R.; Dodsworth, S.; Knapp, S.; Kelly, L.J.; Chase, M.W.; Baldwin, I.T.; et al. The effect of polyploidy and hybridization on the evolution of floral colour in Nicotiana (Solanaceae). Ann. Bot. 2015, 115, 1117–1131. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coefficients | Estimate | SE | t Value | p Value | Tukey HSD | p Value |
|---|---|---|---|---|---|---|
| Intercept | 20.29 | 1.43 | 14.20 | 1.8 × 10−13 | diploid-natural polyploid | 0.0059 |
| Ploidy-natural polyploid | 6.91 | 2.02 | 3.42 | 0.00217 | diploid-synthetic polyploid | 1.0 × 10−6 |
| Ploidy-synthetic polyploid | 12.67 | 1.85 | 6.87 | 3.37 × 10−7 | natural polyploid-synthetic polyploid | 0.012 |
| Coefficients | Estimate | SE | t Value | p Value | Tukey HSD | p Value |
|---|---|---|---|---|---|---|
| Intercept | 609.88 | 25.91 | 23.53 | 2 × 10−16 | diploid-natural polyploid | 0.0059 |
| Ploidy-natural polyploid | −38.13 | 36.65 | −1.04 | 0.30812 | diploid-synthetic polyploid | 1.0 × 10−6 |
| Ploidy-synthetic polyploid | −101.63 | 33.45 | −3.04 | 0.00551 | natural polyploid-synthetic polyploid | 0.012 |
| Coefficients | Estimate | SE | t Value | p Value |
|---|---|---|---|---|
| Intercept | −15.47 | 1.15 | −13.47 | 8.39 × 10−12 |
| Width | 0.57 | 0.03 | 17.67 | 4.39 × 10−14 |
| Ploidy-natural polyploid | 6.86 | 2.48 | 2.76 | 0.01164 |
| Ploidy-synthetic polyploid | 7.01 | 2.81 | 2.50 | 0.02097 |
| Cell number | 0.03 | 0.002 | 15.50 | 5.74 × 10−13 |
| Width:ploidy-natural polyploid | −0.27 | 0.09 | −2.83 | 0.00995 |
| Width:ploidy-synthetic polyploid | −0.28 | 0.09 | −3.12 | 0.00516 |
| Coefficients | Sum Sq | DF | F Value | p Value |
|---|---|---|---|---|
| Intercept | 58.68 | 1 | 181.47 | 8.39 × 10−12 |
| Width | 100.98 | 1 | 312.27 | 4.39 × 10−14 |
| Ploidy | 4.24 | 2 | 6.56 | 0.0061 |
| Cell number | 77.65 | 1 | 240.12 | 5.74 × 10−13 |
| Width:ploidy | 5.07 | 2 | 7.84 | 0.0029 |
| Residuals | 6.79 | 21 |
| Nicotiana sylvestris | |||||||
| Assembler | Data | Number of Contigs | N50 | N90 | % Complete Single Copy | % Fragmented | % Missing |
| SPAdes | Illumina | 44,350 | 1716 | 463 | 78.5 | 7.7 | 13.8 |
| SPAdes | Illumina + ENA + nanopore | 39,620 | 2254 | 760 | 86.8 | 7 | 6.2 |
| SPAdes | ENA + nanopore | 37,097 | 2356 | 788 | 86.4 | 7.6 | 6 |
| Trinity | Illumina | 119,790 | 815 | 336 | 68.9 | 13.4 | 17.7 |
| Trinity | ENA + nanopore * | NA | NA | NA | NA | NA | NA |
| TransAbyss | ENA + nanopore | 100,463 | 1172 | 357 | 84.9 | 7.5 | 7.6 |
| Nicotiana tomentosiformis | |||||||
| Assembler | Data | Number of Contigs | N50 | N90 | % Complete Single Copy | % Fragmented | % Missing |
| SPAdes | Illumina | 47,658 | 1704 | 416 | 79.5 | 7.3 | 13.2 |
| SPAdes | Illumina + ENA + nanopore | 51,360 | 2258 | 643 | 83.6 | 7.7 | 8.7 |
| SPAdes | ENA + nanopore | 39,950 | 2365 | 777 | 82.7 | 8.1 | 9.2 |
| Trinity | Illumina | 132,048 | 788 | 337 | 60.5 | 17.5 | 22 |
| Trinity | ENA + nanopore * | NA | NA | NA | NA | NA | NA |
| TransAbyss | ENA + nanopore | 129,220 | 1068 | 354 | 85.6 | 5.6 | 8.8 |
| Taxon | 60% vs. 85% | 60% vs. 95% | 85% vs. 95% |
|---|---|---|---|
| N. sylvestris | 35/23,952 | 537/23,952 | 0/24,063 |
| N. tomentosiformis | 227/24,585 | 801/24,871 | 0/24,445 |
| N. tabacum 095-55 | 860/43,263 | 3796/44,207 | 691/43,615 |
| N. tabacum ‘Chulumani’ | 2241/43,638 | 4289/43,798 | 1/42,877 |
| synthetic N. tabacum QM24 | 363/42,735 | 2914/42,879 | 262/42,937 |
| synthetic N. tabacum QM25 | 679/43,416 | 2720/43,977 | 335/43,373 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landis, J.B.; Kurti, A.; Lawhorn, A.J.; Litt, A.; McCarthy, E.W. Differential Gene Expression with an Emphasis on Floral Organ Size Differences in Natural and Synthetic Polyploids of Nicotiana tabacum (Solanaceae). Genes 2020, 11, 1097. https://doi.org/10.3390/genes11091097
Landis JB, Kurti A, Lawhorn AJ, Litt A, McCarthy EW. Differential Gene Expression with an Emphasis on Floral Organ Size Differences in Natural and Synthetic Polyploids of Nicotiana tabacum (Solanaceae). Genes. 2020; 11(9):1097. https://doi.org/10.3390/genes11091097
Chicago/Turabian StyleLandis, Jacob B., Amelda Kurti, Amber J. Lawhorn, Amy Litt, and Elizabeth W. McCarthy. 2020. "Differential Gene Expression with an Emphasis on Floral Organ Size Differences in Natural and Synthetic Polyploids of Nicotiana tabacum (Solanaceae)" Genes 11, no. 9: 1097. https://doi.org/10.3390/genes11091097
APA StyleLandis, J. B., Kurti, A., Lawhorn, A. J., Litt, A., & McCarthy, E. W. (2020). Differential Gene Expression with an Emphasis on Floral Organ Size Differences in Natural and Synthetic Polyploids of Nicotiana tabacum (Solanaceae). Genes, 11(9), 1097. https://doi.org/10.3390/genes11091097