Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution


Abstract
1. Introduction
2. Materials and Methods
2.1. Transposable Elements Reference Construction
2.2. Identification and Annotation of Transposable Elements
2.3. Transposable Element Loads and Expansion Analyses
2.4. Reconstructing the Phylogenetic Tree
2.5. Time-Calibrated Phylogeny
2.6. Fitting Multiple Phylogenetic Comparative Models
2.7. Phylogenetically Independent Contrasts
2.8. Ancestral State Reconstruction of Transposable Element Loads
2.9. Identifying Shifts of Transposable Element Loads Change Rates in the Phylogeny
2.10. Transposable Element Protein Annotation
2.11. The Phylogenetic Analyses of LTRs in LEPIDOPTERA
2.12. Horizontal Transposable Element Transfer
2.13. Analysis of Transposable Elements Distribution in Arthropods
2.14. The ArTEdb Database
3. Results
3.1. Construction of Transposable Element References
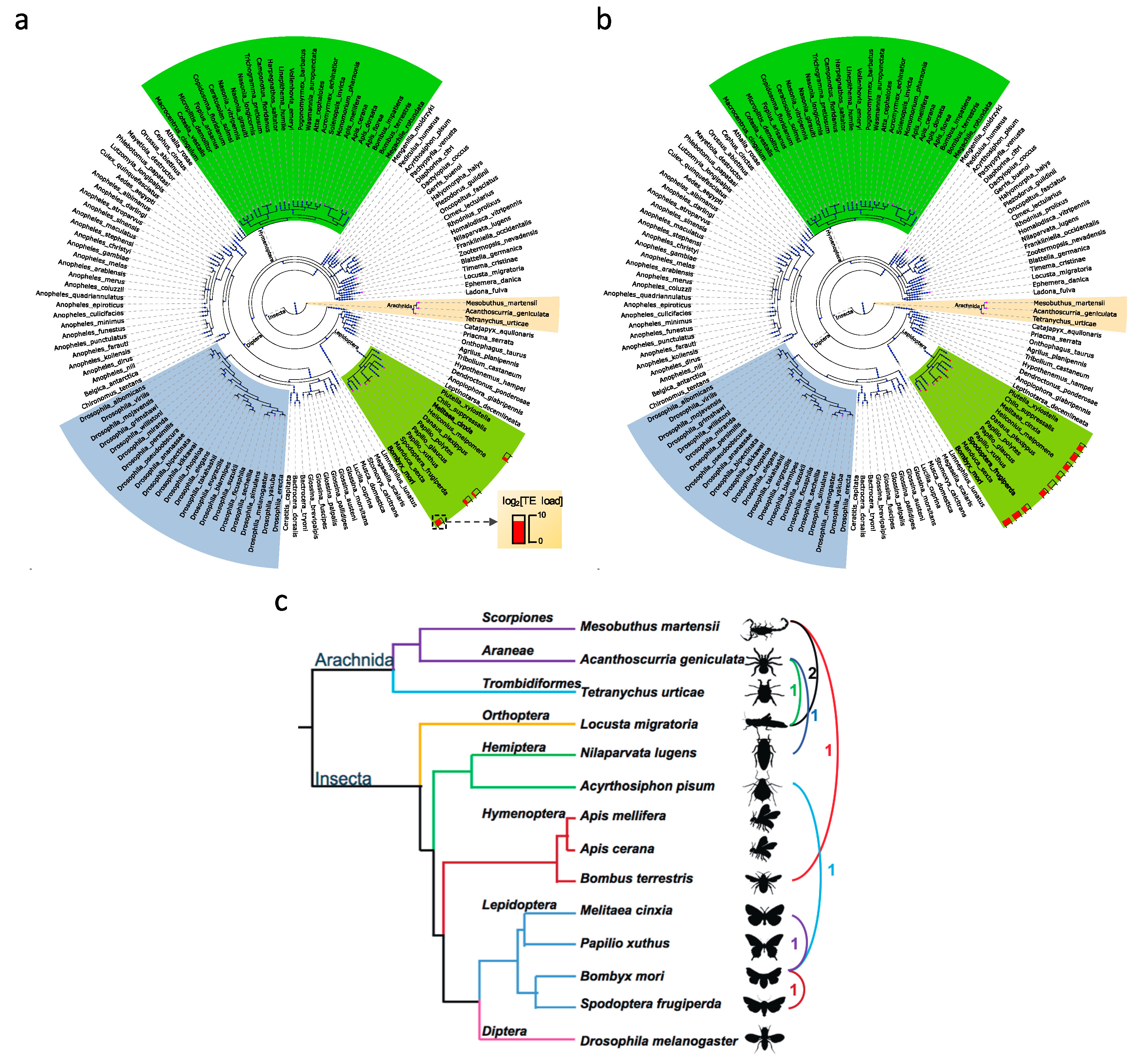
3.2. Transposable Element Loads Vary Greatly in Arthropods
3.2.1. Transposable Element Loss is Prevalent in Hymenoptera
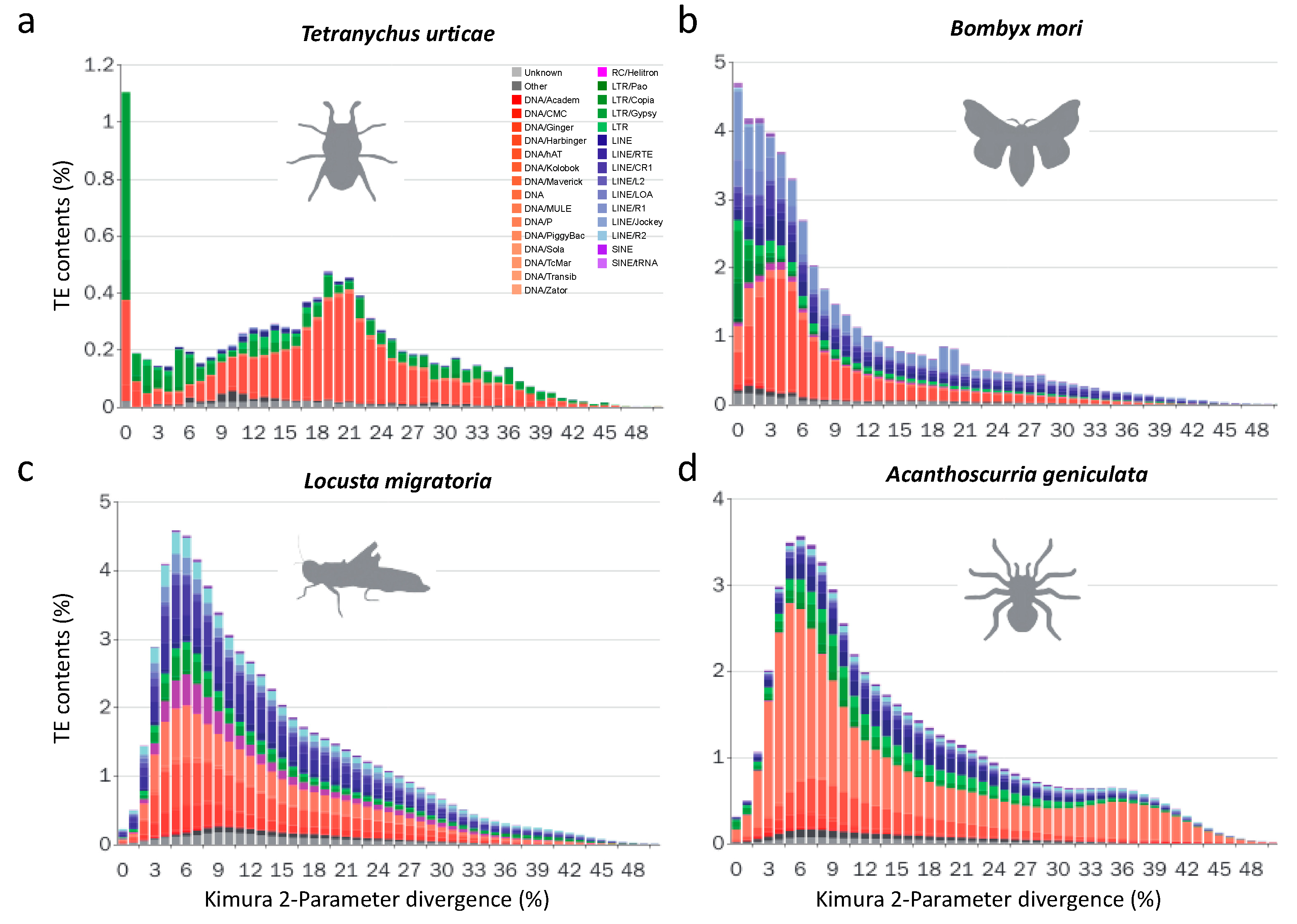
3.2.2. Recent LTR Expansion in T. urticae
3.2.3. Lepidopterans Have Diversified Transposable Elements Subfamilies and Large Transposable Elements Loads
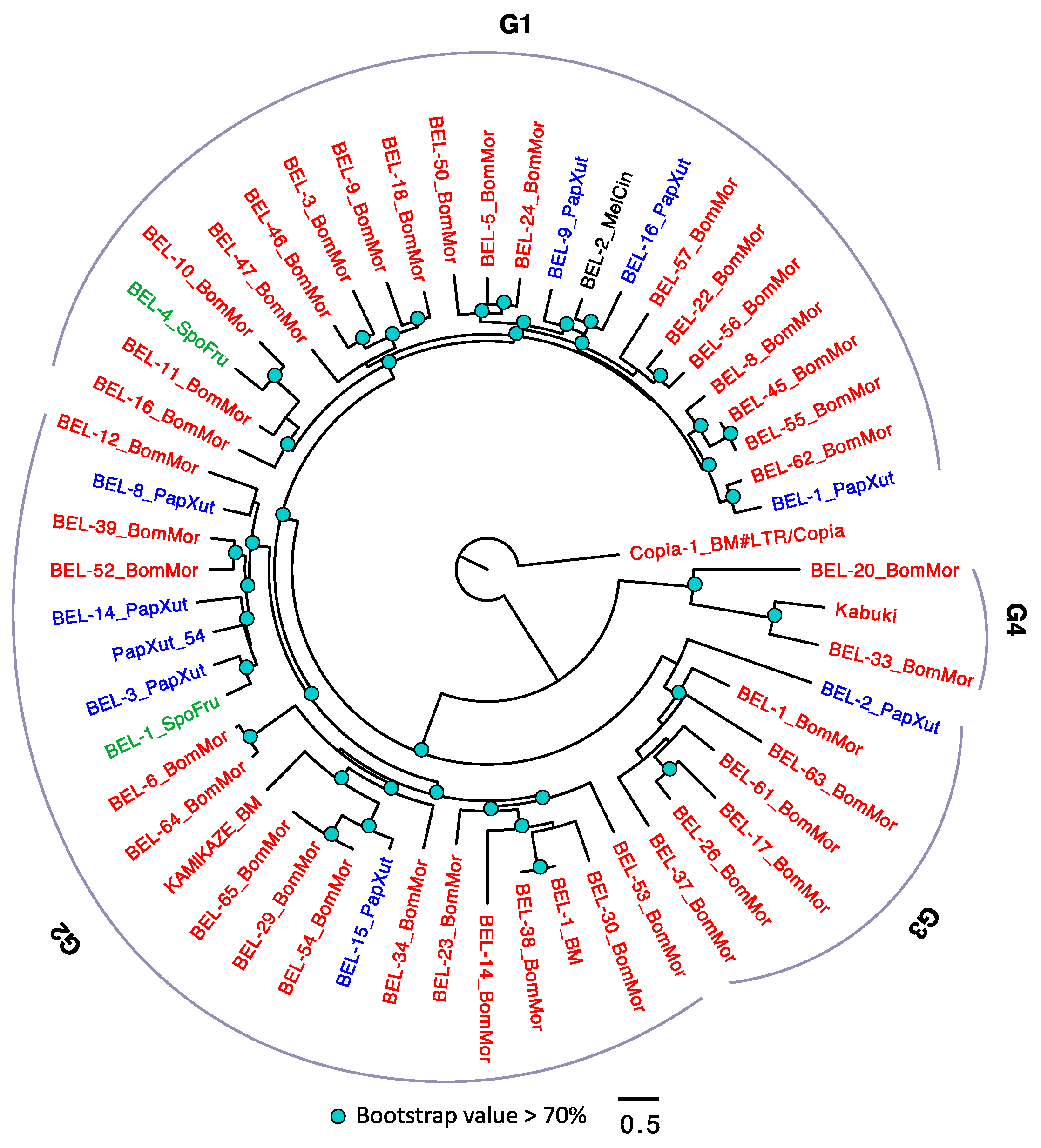
3.2.4. The Evolutionary History of LTRs in Lepidoptera
3.2.5. Non-LTR Transposable Elements Contribute More to Arthropods with Larger Genomes
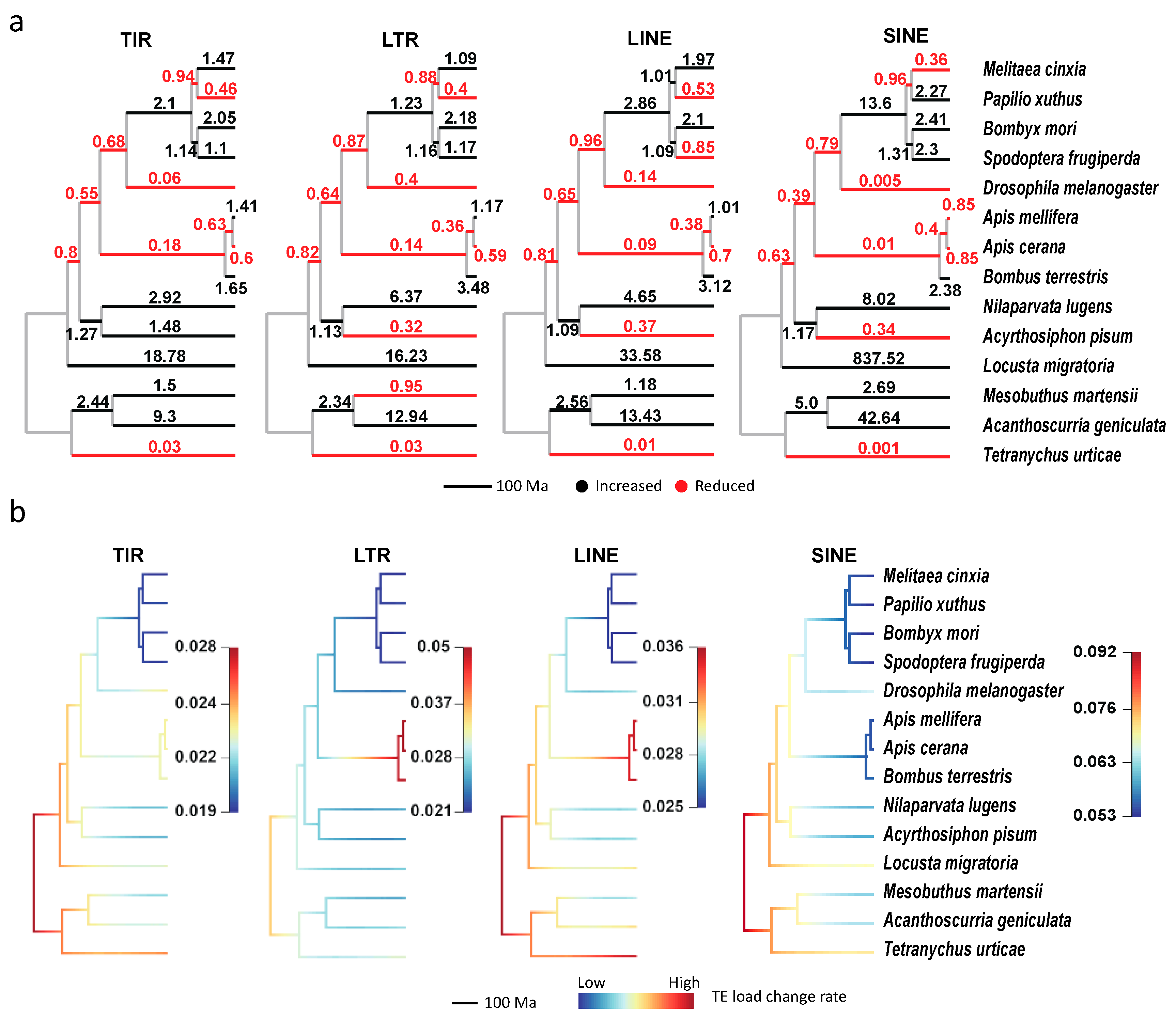
3.3. The Expansion and Contraction of Transposable Elements in Arthropods
3.4. Transposable Element Loads are Significantly Correlated with Genome Sizes
3.5. Horizontal Transposable Element Transfer in Arthropods
3.6. The ArTEdb
4. Discussion
4.1. A New Database of High-Quality Transposable Element References for Arthropods
4.2. Why Does the Transposable Element Loads Can Be So Different Across Arthropod Species?
4.3. The Contribution of HTTs to the Transposable Element Repertoire in Arthropod Genomes
4.4. Adaptive Transposable Element Insertions in Arthropods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E.; Crick, F. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D. The Genome Sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Ahola, V.; Lehtonen, R.J.; Somervuo, P.; Salmela, L.; Koskinen, P.; Rastas, P.; Välimäki, N.; Paulin, L.; Kvist, J.; Wahlberg, N.; et al. The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat. Commun. 2014, 5, 4737. [Google Scholar] [PubMed]
- Cao, Z.; Yu, Y.; Wu, Y.; Hao, P.; Di, Z.; He, Y.; Chen, Z.; Yang, W.; Shen, Z.; He, X.; et al. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 2013, 4, 2602. [Google Scholar] [CrossRef]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; De Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef]
- Grbić, M.; Van Leeuwen, T.; Clark, R.M.; Rombauts, S.; Rouzé, P.; Grbić, V.; Osborne, E.J.; Dermauw, W.; Ngoc, P.C.T.; Ortego, F.; et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 2011, 479, 487–492. [Google Scholar] [CrossRef] [PubMed]
- International Aphid Genomics Consortium. Genome sequence of the pea aphid acyrthosiphon pisum. PLoS Biol. 2010, 8, e1000313. [Google Scholar]
- International Aphid Genomics Consortium. The genome of a lepidopteran model insect, the silkworm bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Kakumani, P.K.; Malhotra, P.; Mukherjee, S.K.; Bhatnagar, R.K. A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics 2014, 104, 134–143. [Google Scholar] [CrossRef]
- Li, X.; Fan, D.; Zhang, W.; Liu, G.; Zhang, L.; Zhao, L.; Fang, X.; Chen, L.; Dong, Y.; Chen, Y.; et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 2015, 6, 8212. [Google Scholar] [CrossRef]
- Park, D.; Jung, J.W.; Choi, B.-S.; Jayakodi, M.; Lee, J.; Lim, J.; Yu, Y.; Choi, Y.-S.; Lee, M.-L.; Park, Y.; et al. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genom. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Sadd, B.M.; Barribeau, S.M.; Bloch, G.; De Graaf, D.C.; Dearden, P.; Elsik, C.G.; Gadau, J.; Grimmelikhuijzen, C.J.; Hasselmann, M.; Lozier, J.D.; et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.; Cheng, L.; Fan, D.; Feng, Y.; et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 3765. [Google Scholar] [CrossRef]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5, 2957. [Google Scholar] [CrossRef]
- Xue, J.; Zhou, X.; Zhang, C.X.; Yu, L.L.; Fan, H.W.; Wang, Z.; Xu, H.J.; Xi, Y.; Zhu, Z.R.; Zhou, W.W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Armisén, D.; Gibbs, R.A.; Hering, L.; Khila, A.; Mayer, G.; Richards, S.; Niehuis, O.; Misof, B. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Evol. Biol. 2019, 19, 11. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet. Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [PubMed]
- Sessegolo, C.; Burlet, N.; Haudry, A. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol. Lett. 2016, 12, 20160407. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The gypsy database (gydb) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R.; Avni, D.; Biberman, Y.; Meyuhas, O. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R.; Mcgettigan, P.; Mc William, H.; Valentin, F.; Wallace, I.; Wilm, A.; López, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0 2013–2015. Available online: http://www.repeatmasker.org (accessed on 24 April 2017).
- Hoede, C.; Arnoux, S.; Moisset, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An Automatic Transposable Element Classification Tool. PLoS ONE 2014, 9, 91929. [Google Scholar] [CrossRef]
- Abrusán, G.; Grundmann, N.; Demester, L.; Makalowski, W. TEclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Haudry, A.; Lerat, E. “One code to find them all”: A perl tool to conveniently parse repeatmasker output files. Mob. DNA 2014, 5, 13. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2017, 35, 543–548. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Wernersson, R. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Rehm, P.; Borner, J.; Meusemann, K.; Von Reumont, B.M.; Simon, S.; Hadrys, H.; Misof, B.; Burmester, T. Dating the arthropod tree based on large-scale transcriptome data. Mol. Phylo. Evol. 2011, 61, 880–887. [Google Scholar] [CrossRef]
- Revell, L.J. Phytools: An r package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Pennell, M.W.; Eastman, J.M.; Slater, G.J.; Brown, J.; Uyeda, J.C.; Fitzjohn, R.G.; Alfaro, M.E.; Harmon, L.J. geiger v2.0: An expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics 2014, 30, 2216–2218. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Maximum-likelihood estimation of evolutionary trees from continuous characters. Am. J. Hum. Genet. 1973, 25, 471–492. [Google Scholar]
- Butler, M.A.; King, A.A. Phylogenetic Comparative Analysis: A Modeling Approach for Adaptive Evolution. Am. Nat. 2004, 164, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Harmon, L.J.; Losos, J.B.; Davies, T.J.; Gillespie, R.G.; Gittleman, J.L.; Jennings, W.B.; Kozak, K.H.; McPeek, M.; Moreno-Roark, F.; Near, T.J.; et al. Early bursts of body size and shape evolution are rare in comparative data. Evolution 2010, 64, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Statist. 1979, 6, 65–70. [Google Scholar]
- Pagel, M. Detecting correlated evolution on phylogenies: A general method for the comparative analysis of discrete characters. Proc. R. Soc. Lond. B. 1994, 255, 37–45. [Google Scholar]
- Szitenberg, A.; Cha, S.; Opperman, C.H.; Bird, D.M.; Blaxter, M.L.; Lunt, D.H. Genetic Drift, Not Life History or RNAi, Determine Long-Term Evolution of Transposable Elements. Genome Biol. Evol. 2016, 8, 2964–2978. [Google Scholar] [CrossRef]
- Szitenberg, A.; John, M.; Blaxter, M.L.; Lunt, D.H. ReproPhylo: An Environment for Reproducible Phylogenomics. PLoS Comput. Biol. 2015, 11, e1004447. [Google Scholar] [CrossRef] [PubMed]
- Rabosky, D.L.; Donnellan, S.C.; Grundler, M.; Lovette, I.J.; West, C.; James, S.A.; Davey, R.P.; Dicks, J.; Roberts, I.N. Analysis and Visualization of Complex Macroevolutionary Dynamics: An Example from Australian Scincid Lizards. Syst. Biol. 2014, 63, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Rabosky, D.L.; Goldberg, E.E. Model Inadequacy and Mistaken Inferences of Trait-Dependent Speciation. Syst. Biol. 2015, 64, 340–355. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Santini, F.; Eastman, J.; Smith, S.A.; Sidlauskas, B.; Chang, J.; Alfaro, M.E. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nat. Commun. 2013, 4, 1958. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Gründler, M.; Anderson, C.; Title, P.; Shi, J.J.; Brown, J.W.; Huang, H.; Larson, J.G. BAMMtools: An R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 2014, 5, 701–707. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Thomas, G.W.C.; Dohmen, E.; Hughes, D.S.T.; Murali, S.C.; Poelchau, M.; Glastad, K.; Anstead, C.A.; Ayoub, N.A.; Batterham, P.; Bellair, M.; et al. The genomic basis of arthropod diversity. bioRxiv 2018, 382945. [Google Scholar]
- Pearson, W.R. Rapid and sensitive sequence comparison with FASTP and FASTA. Meth. Enzymol. 1990, 183, 63–98. [Google Scholar]
- Misra, S.; Crosby, M.; Mungall, C.J.; Matthews, B.B.; Campbell, K.S.; Hradecky, P.; Huang, Y.; Kaminker, J.S.; Millburn, G.H.; Prochnik, S.; et al. Annotation of the Drosophila melanogaster euchromatic genome: A systematic review. Genome Biol. 2002, 3, 83. [Google Scholar] [CrossRef]
- Quesneville, H.; Bergman, C.M.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabéhère, D.; Stormo, G. Combined Evidence Annotation of Transposable Elements in Genome Sequences. PLoS Comput. Biol. 2005, 1, e22. [Google Scholar] [CrossRef]
- Xu, H.-E.; Zhang, H.-H.; Xia, T.; Han, M.-J.; Shen, Y.-H.; Zhang, Z. BmTEdb: A collective database of transposable elements in the silkworm genome. Database 2013, 2013, 55. [Google Scholar] [CrossRef]
- Obbard, D.J.; Gordon, K.H.; Buck, A.H.; Jiggins, F.M. The evolution of rnai as a defence against viruses and transposable elements. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 99–115. [Google Scholar] [CrossRef]
- Zhao, X.; Xiong, J.; Mao, F.; Sheng, Y.; Chen, X.; Feng, L.; Dui, W.; Yang, W.; Kapusta, A.; Feschotte, C.; et al. RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Genes Dev. 2019, 33, 348–364. [Google Scholar] [CrossRef]
- Revell, L.J.; Harmon, L.J.; Collar, D.C. Phylogenetic Signal, Evolutionary Process, and Rate. Syst. Biol. 2008, 57, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G. Transposable elements and the evolution of genome size in eukaryotes. Genetica 2002, 115, 49–63. [Google Scholar] [CrossRef]
- Felsenstein, J. Phylogenies and the Comparative Method. Am. Nat. 1985, 125, 1–15. [Google Scholar] [CrossRef]
- Daniels, S.B.; Peterson, K.R.; Strausbaugh, L.D.; Kidwell, M.G.; Chovnick, A. Evidence for Horizontal Transmission of the P Transposable Element between Drosophila Species. Genetics 1990, 124, 339–355. [Google Scholar]
- Bingham, P.M.; Kidwell, M.G.; Rubin, G.M. The molecular basis of P-M hybrid dysgenesis: The role of the P element, a P-strain-specific transposon family. Cell 1982, 29, 995–1004. [Google Scholar] [CrossRef]
- Gilbert, C.; Feschotte, C. Horizontal acquisition of transposable elements and viral sequences: Patterns and consequences. Curr. Opin. Genet. Dev. 2018, 49, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Peccoud, J.; Loiseau, V.; Cordaux, R.; Gilbert, C. Massive horizontal transfer of transposable elements in insects. Proc. Natl. Acad. Sci. USA 2017, 114, 4721–4726. [Google Scholar] [CrossRef] [PubMed]
- Blumenstiel, J.P. Evolutionary dynamics of transposable elements in a small RNA world. Trends Genet. 2011, 27, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Reiss, D.; Mialdea, G.; Miele, V.; De Vienne, D.M.; Peccoud, J.; Gilbert, C.; Duret, L.; Charlat, S. Global survey of mobile DNA horizontal transfer in arthropods reveals Lepidoptera as a prime hotspot. PLoS Genet. 2019, 15, e1007965. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Loreto, E.L.; Clark, J.B. Factors that affect the horizontal transfer of transposable elements. Curr. Issues Mol. Biol. 2004, 6, 57–71. [Google Scholar] [PubMed]
- Goerner-Potvin, P.; Bourque, G. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018, 19, 688–704. [Google Scholar] [CrossRef]
- Pasyukova, E.G.; Nuzhdin, S.V.; Morozova, T.V.; Mackay, T.F.C. Accumulation of Transposable Elements in the Genome of Drosophila melanogaster is Associated with a Decrease in Fitness. J. Hered. 2004, 95, 284–290. [Google Scholar] [CrossRef]
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef]
- Bewick, A.J.; Vogel, K.J.; Moore, A.J.; Schmitz, R.J. Evolution of DNA methylation across insects. Mol. Biol. Evol. 2017, 34, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.H.; Quarles, K.A.; Yang, Y.; Tanguy, M.; Frezal, L.; Smith, S.A.; Sharma, P.P.; Cordaux, R.; Gilbert, C.; Giraud, I.; et al. Pan-arthropod analysis reveals somatic pirnas as an ancestral defence against transposable elements. Nat. Ecol. Evol. 2018, 2, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Clark, A.G. Population dynamics of PIWI-interacting RNAs (piRNAs) and their targets in Drosophila. Genome Res. 2009, 20, 212–227. [Google Scholar] [CrossRef]
- El Baidouri, M.; Cooke, R.; Gao, D.; Lasserre, E.; Llauro, C.; Mirouze, M.; Picault, N.; Panaud, O.; Carpentier, M.-C.; Jackson, S. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 2014, 24, 831–838. [Google Scholar] [CrossRef]
- Drezen, J.-M.; Josse, T.; Bézier, A.; Gauthier, J.; Huguet, E.; Herniou, E.A. Impact of Lateral Transfers on the Genomes of Lepidoptera. Genes 2017, 8, 315. [Google Scholar] [CrossRef]
- Gilbert, C.; Peccoud, J.; Chateigner, A.; Moumen, B.; Cordaux, R.; Herniou, E.A. Continuous Influx of Genetic Material from Host to Virus Populations. PLoS Genet. 2016, 12, e1005838. [Google Scholar] [CrossRef]
- Luo, S.; Lu, J. Silencing of Transposable Elements by piRNAs in Drosophila: An Evolutionary Perspective. Genom. Proteom. Bioinform. 2017, 15, 164–176. [Google Scholar] [CrossRef]
- Yamashiro, H.; Siomi, M.C. Piwi-interacting RNA in drosophila: Biogenesis, transposon regulation, and beyond. Chem. Rev. 2018, 118, 4404–4421. [Google Scholar] [CrossRef]
- Bartolomé, C.; Bello, X.; Maside, X. Widespread evidence for horizontal transfer of transposable elements across Drosophila genomes. Genome Biol. 2009, 10, R22. [Google Scholar] [CrossRef]
- Nigumann, P.; Redik, K.; Mätlik, K.; Speek, M. Many Human Genes Are Transcribed from the Antisense Promoter of L1 Retrotransposon. Genomics 2002, 79, 628–634. [Google Scholar] [CrossRef]
- Van De Lagemaat, L.N.; Landry, J.-R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef]
- Chen, B.; Shilova, V.Y.; Zatsepina, O.G.; Evgen’Ev, M.B.; Feder, M.E. Location of P element insertions in the proximal promoter region of Hsp70A is consequential for gene expression and correlated with fecundity in Drosophila melanogaster. Cell Stress Chaperones 2008, 13, 11–17. [Google Scholar] [CrossRef]
- Bejerano, G.; Lowe, C.B.; Ahituv, N.; King, B.; Siepel, A.; Salama, S.R.; Rubin, E.M.; Kent, W.J.; Haussler, D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 2006, 441, 87–90. [Google Scholar] [CrossRef]
- Smith, A.M.; Sanchez, M.-J.; Follows, G.A.; Kinston, S.; Donaldson, I.J.; Green, A.R.; Gottgens, B. A novel mode of enhancer evolution: The Tal1 stem cell enhancer recruited a MIR element to specifically boost its activity. Genome Res. 2008, 18, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.F.; Lopez-Leal, R.; Nasif, S.; Beati, P.; Gelman, D.M.; Low, M.J.; de Souza, F.J.; Rubinstein, M. Convergent evolution of two mammalian neuronal enhancers by sequential exaptation of unrelated retroposons. Proc. Natl. Acad. Sci. USA 2011, 108, 15270–15275. [Google Scholar] [CrossRef]
- Bundock, P.; Hooykaas, P. An Arabidopsis hAT-like transposase is essential for plant development. Nature 2005, 436, 282–284. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. RAG1 Core and V(D)J Recombination Signal Sequences Were Derived from Transib Transposons. PLoS Biol. 2005, 3, e181. [Google Scholar] [CrossRef] [PubMed]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]
- De Souza, F.S.; Franchini, L.F.; Rubinstein, M. Exaptation of Transposable Elements into Novel Cis-Regulatory Elements: Is the Evidence Always Strong? Mol. Biol. Evol. 2013, 30, 1239–1251. [Google Scholar] [CrossRef]
- Joly-Lopez, Z.; Bureau, T. Exaptation of transposable element coding sequences. Curr. Opin. Genet. Dev. 2018, 49, 34–42. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Aminetzach, Y.T.; MacPherson, J.M.; Petrov, D.A. Pesticide Resistance via Transposition-Mediated Adaptive Gene Truncation in Drosophila. Science 2005, 309, 764–767. [Google Scholar] [CrossRef]
- Hof, A.E.V.; Campagne, P.; Rigden, D.J.; Yung, C.J.; Lingley, J.; Quail, M.A.; Hall, N.; Darby, A.C.; Saccheri, I.J. The industrial melanism mutation in British peppered moths is a transposable element. Nature 2016, 534, 102–105. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Order | Species | Genome Sizes † | TE Reference Sequences | ||
|---|---|---|---|---|---|---|
| RB/RM/LTR | RTE/DTE/UnC | Total | ||||
| Insecta | Diptera | Drosophila melanogaster | 144 Mb | 147/79/8 | 159/47/28 | 234 |
| Lepidoptera | Spodoptera frugiperda | 358 Mb | 1/475/12 | 263/190/35 | 488 | |
| Bombyx mori‡ | 460 Mb | 92/546/183 | 552/232/37 | 821 | ||
| Papilio xuthus | 244 Mb | 41/319/60 | 248/151/21 | 420 | ||
| Melitaea cinxia | 390 Mb | 0/763/17 | 433/299/48 | 780 | ||
| Hymenoptera | Bombus terrestris | 249 Mb | 6/520/12 | 241/267/30 | 538 | |
| Apis cerana | 228 Mb | 0/86/1 | 28/49/10 | 87 | ||
| Apis mellifera | 250 Mb | 6/136/1 | 35/98/10 | 143 | ||
| Hemiptera | Acyrthosiphon pisum | 542 Mb | 331/752/75 | 326/796/36 | 1158 | |
| Nilaparvata lugens | 1.14 Gb | 0/1,136/230 | 872/419/75 | 1366 | ||
| Orthoptera | Locusta migratoria | 5.6 Gb | 1028/1182/56 | 1144/1018/104 | 2266 | |
| Arachnida | Trombidiformes | Tetranychus urticae | 90 Mb | 10/122/73 | 105/86/14 | 205 |
| Araneae | Acanthoscurria geniculata | 7.2 Gb | 0/1857/167 | 967/982/75 | 2024 | |
| Scorpiones | Mesobuthus martensii | 925 Mb | 39/1260/101 | 476/843/81 | 1400 | |
| TE Subfamilies | Correlation Coefficient | P | Corrected P |
|---|---|---|---|
| LINE/L2 | 0.868 | 1.20 × 10−4 | 3.01 × 10−3 |
| SINE/Unclassified | 0.784 | 1.51 × 10−3 | 3.18 × 10−2 |
| LINE/Unclassified | 0.760 | 2.55 × 10−3 | 4.85 × 10−2 |
| DNA/Unclassified | 0.756 | 2.79 × 10−3 | 5.02 × 10−2 |
| DNA/TcMar | 0.728 | 4.75 × 10−3 | 8.07 × 10−2 |
| LTR/Copia | 0.724 | 5.14 × 10−3 | 8.22 × 10−2 |
| DNA/hAT | 0.654 | 1.53 × 10−2 | 2.13 × 10−1 |
| LINE/CR1 | 0.644 | 1.75 × 10−2 | 2.13 × 10−1 |
| LINE/RTE | 0.652 | 1.57 × 10−2 | 2.13 × 10−1 |
| LTR/Unclassified | 0.654 | 1.52 × 10−2 | 2.13 × 10−1 |
| LTR/Pao | 0.644 | 1.76 × 10−2 | 2.13 × 10−1 |
| DNA/Ginger | 0.559 | 4.72 × 10−2 | 4.25 × 10−1 |
| LINE/I | 0.511 | 7.45 × 10−2 | 5.96 × 10−1 |
| DNA/CMC | 0.489 | 8.97 × 10−2 | 6.28 × 10−1 |
| DNA/Academ | 0.419 | 1.54 × 10−1 | 9.23 × 10−1 |
| LINE/Dong-R4 | 0.403 | 1.73 × 10−1 | 9.23 × 10−1 |
| DNA/Harbinger | 0.358 | 2.30 × 10−1 | 9.23 × 10−1 |
| DNA/PiggyBac | 0.371 | 2.12 × 10−1 | 9.23 × 10−1 |
| LINE/Jockey | 0.394 | 1.83 × 10−1 | 9.23 × 10−1 |
| LINE/R1 | 0.351 | 2.40 × 10−1 | 9.23 × 10−1 |
| LTR/Gypsy | 0.382 | 1.98 × 10−1 | 9.23 × 10−1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Lu, J. Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution. Genes 2019, 10, 338. https://doi.org/10.3390/genes10050338
Wu C, Lu J. Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution. Genes. 2019; 10(5):338. https://doi.org/10.3390/genes10050338
Chicago/Turabian StyleWu, Changcheng, and Jian Lu. 2019. "Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution" Genes 10, no. 5: 338. https://doi.org/10.3390/genes10050338
APA StyleWu, C., & Lu, J. (2019). Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution. Genes, 10(5), 338. https://doi.org/10.3390/genes10050338