Biology and Physics of Heterochromatin-Like Domains/Complexes



Abstract
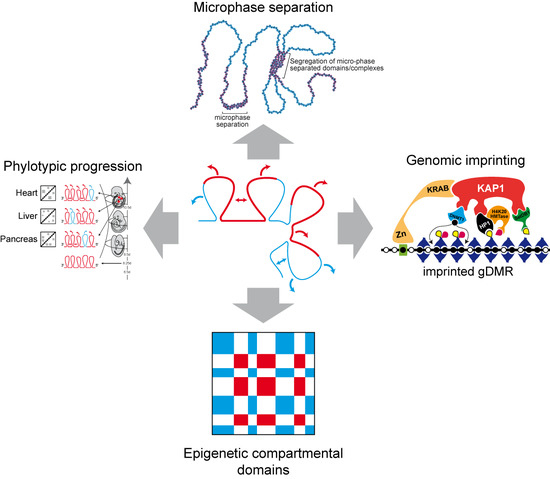
1. Introduction
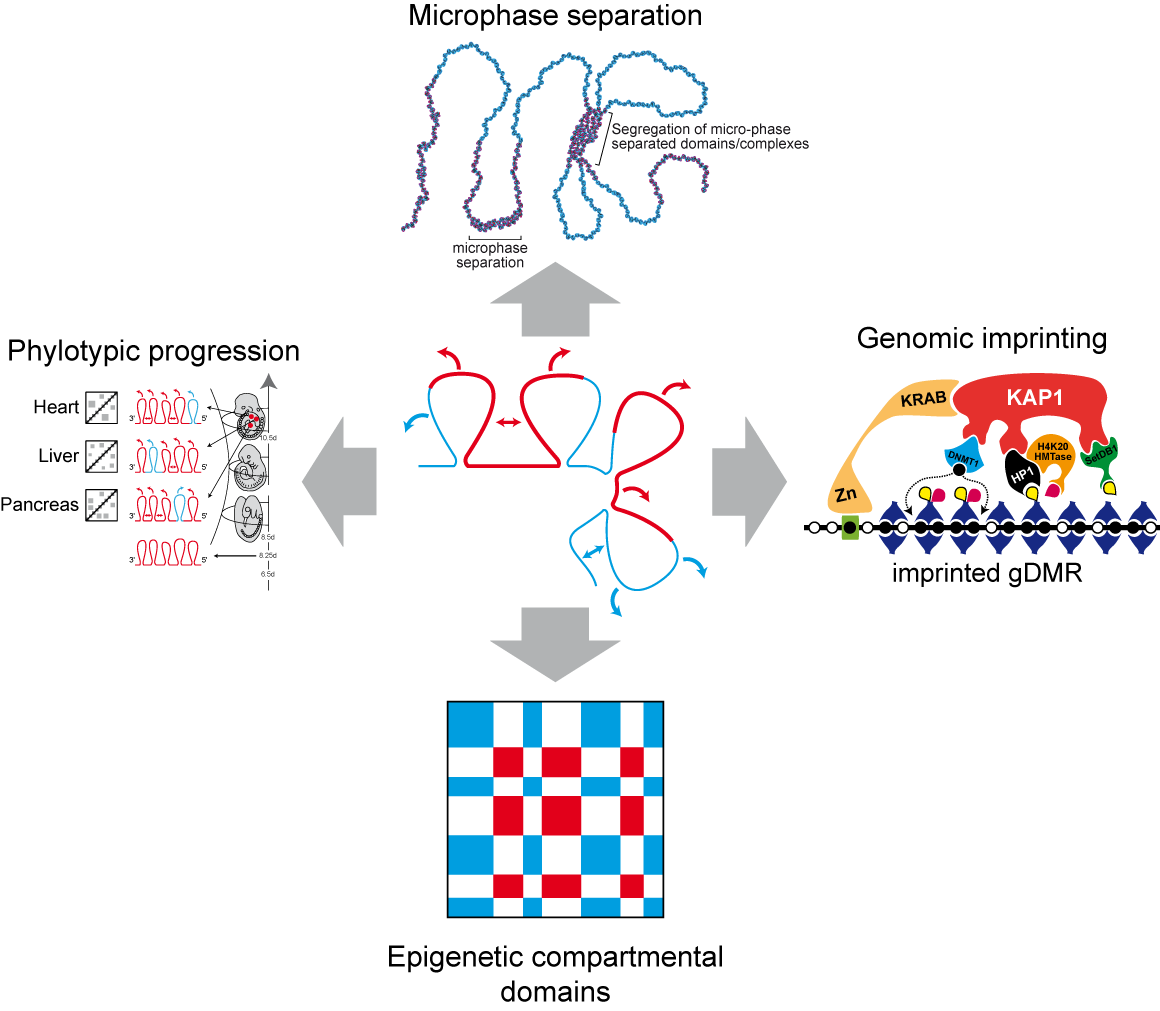
2. Heterochromatin-Like Domains/Complexes in Eukaryotes
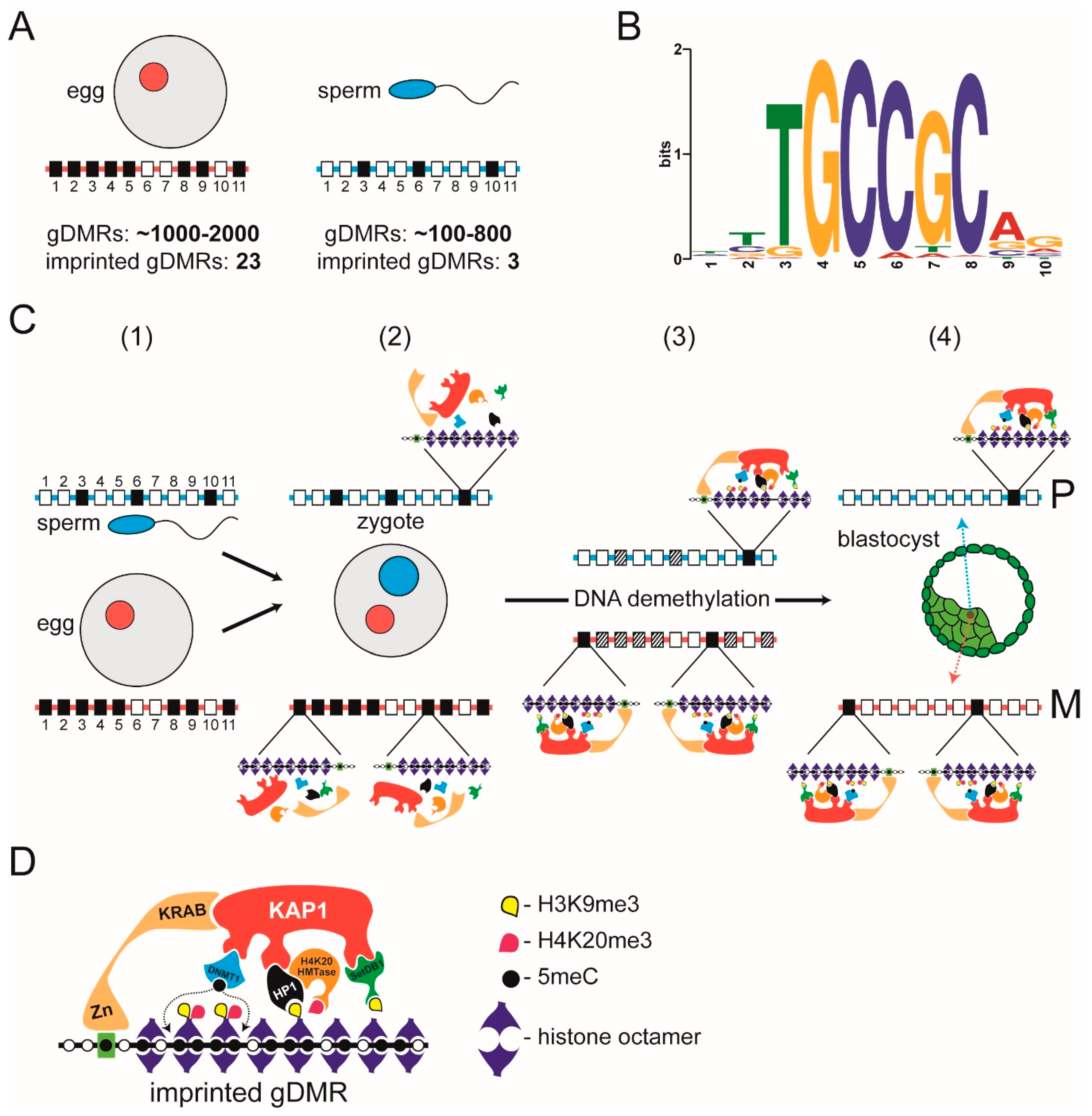
3. Heterochromatin-Like Complexes and Preservation of DNA Methylation at Imprinted gDMRs during Pre-Implantation Embryogenesis
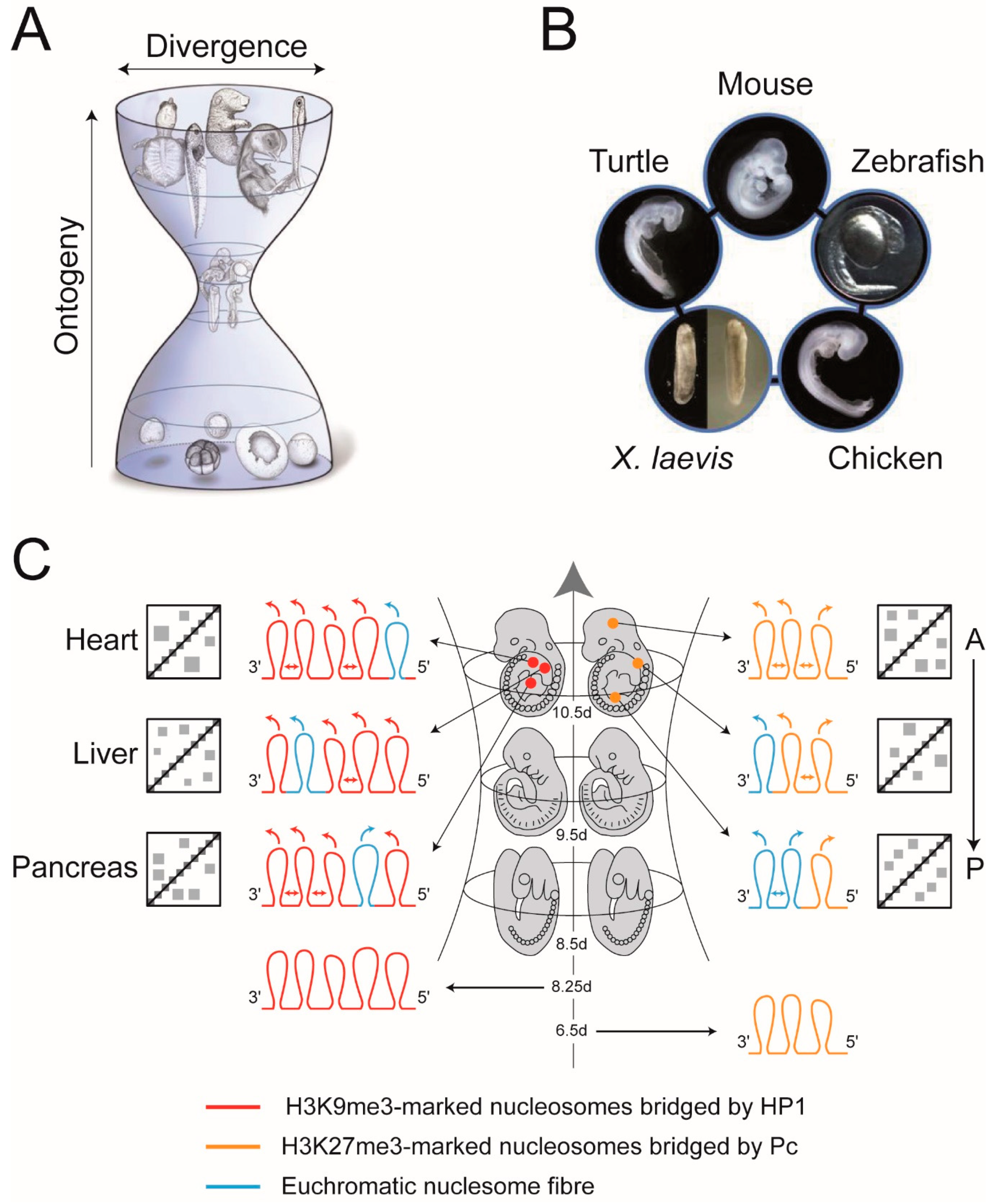
4. Heterochromatin-Like Domains/Complexes and the Phylotypic Progression during Vertebrate Development
5. Heterochromatin-Like Domains/Complexes and Block Copolymers (BCPs)
6. Di-BCPs, χ and The Monomer as the “Unit of Incompatibility”
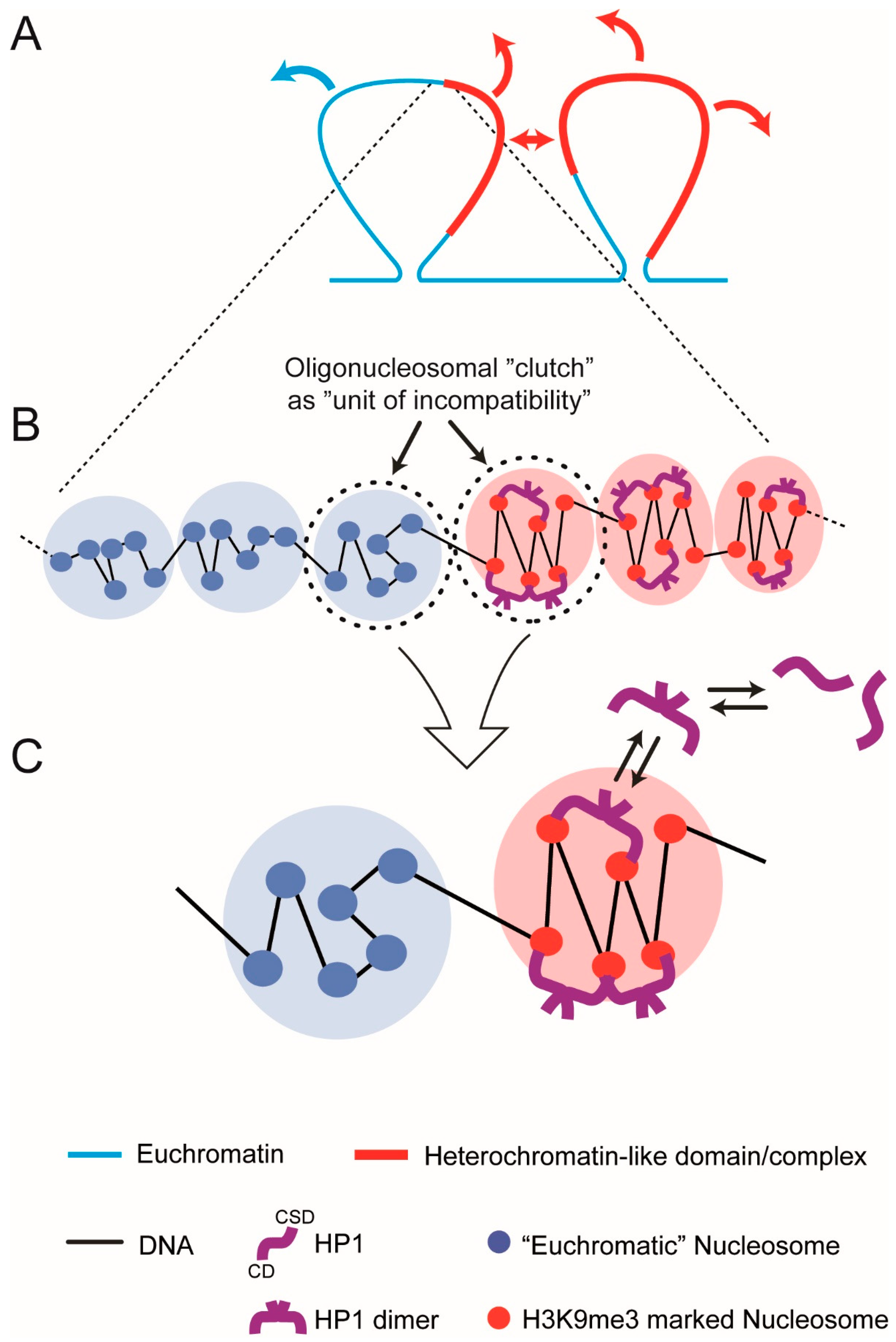
7. The Oligo-Nucleosomal “Clutch” As the “Unit of Incompatibility” of Chromatin
8. Theoretical “Clutch” Model
- A symmetric pair of “clutches”, a euchromatic “clutch” and heterochromatin-like “clutch” (HC) that are connected by linker DNA. Each clutch contains six nucleosomes, a value that lies in the middle of the range 2–10. The HC consisting of H3K9me3-marked nucleosomes represents the repeating unit (“unit of incompatibility”) of a heterochromatin-like domain/complex; likewise, for the euchromatic clutch and euchromatin. Both clutches are equally miscible in the nucleoplasm (a concentrated solution of chromatin fibres) reducing the problem to incompatibility between the clutches.
- In the absence of HP1, the “clutches” are thermodynamically equivalent. Incompatibility between the HC and euchromatic “clutch” results from “bridging” of two K9me3 motifs on histone H3 proteins in separate nucleosomes by HP1 dimers; stacked nucleosomes are the preferred HP1 binding site. Bridging is maintained by constant exchange of bound HP1 dimers with free dimers in the nucleoplasm.
- HP1 “bridging” stabilises the zig-zag geometry of H3K9me3-marked nucleosomes and leads to compaction. Nucleosomes in the euchromatic “clutch” are disorganised with limited (unstable) zig-zag geometry.
Characteristics and Caveats of the Theoretical “Clutch” Model
- In the form presented the model may not be applicable to macro-phase separation of cytologically-visible constitutive heterochromatin. There are additional factors that may need to be included that are together severally necessary and jointly sufficient in causing macro-phase separation of constitutive heterochromatin. They include repetitive DNAs, ncRNAs, proteins that bind (modified) DNA/histones [213]. Notably, there is a documented interaction of Swi6HP1 CSD dimer with the H2Bα1 helix that results in the deformation of the nucleosome core that is thought to drive phase separation of constitutive heterochromatin in fission yeast [55]. HP1 also binds to methylated lysine 26 of histone H1.4 (H1.4K26me) in constitutive heterochromatin [51], which may be significant in the context of the observation that H1-containing chromatin preferentially phase separates [210]. These factors are not included in our model since we are concerned with micro-phase separation of heterochromatin-like domains/complexes outside constitutive heterochromatin.
- H3K9me3 epigenetic modification acts as an HP1 binding site only. Absent HP1 an H3K9me3-marked “clutch” is thermodynamically equivalent to the euchromatic “clutch”. Micro-phase separation results only after HP1 “bridging” between H3K9me3-marked nucleosomes. Experimentally, a reference “clutch” consisting of six H3K9me3-marked nucleosomes could be used to measure changes in free energy and excess entropy generated after addition of HP1 to the system. The H3K9me3-marked clutch would need to be tethered to mimic the connection through linker DNA to flanking euchromatic “clutches”.
- HP1 dimer binding is modelled as “bridging” two H3K9-methylated histone H3 molecules in separate nucleosomes [37]. Accordingly, a nucleosome chain with stacked nucleosomes would be the preferred HP1 binding sites [214], which would be consistent with HP1 binding to a fibre with nucleosomes having a zig-zag geometry. Compaction would be promoted by allosteric cooperativity arising from changes in di-nucleosome conformation after HP1 bridging that would in turn enhance further HP1 bridging [214]. No direct HP1-HP1 co-operative binding is assumed, although there is evidence that such cooperative binding might contribute to “spreading” of the heterochromatic state [215,216]. This has been modelled previously [212] and could be incorporated as a free energy contribution (HHP1-HP1)HC to Equation (5).
- Histone acetylation is not included in the model. Histone acetylation is an epigenetic modification generally associated with euchromatin and is known to neutralise the positive charge on the histone tails [217]. Inclusion of histone acetylation could affect the incompatibility between the euchromatic and heterochromatin-like clutches. Our reasoning for not including histone acetylation is that H3K9me3 and H3K27me3 are truly epigenetic and define ECDs (Box S1). They are associated with “write-and-read” activities that ensure their inheritance from one cell generation to the next [218]. Histone acetylation does not possess an analogous write-and-read activity [218].
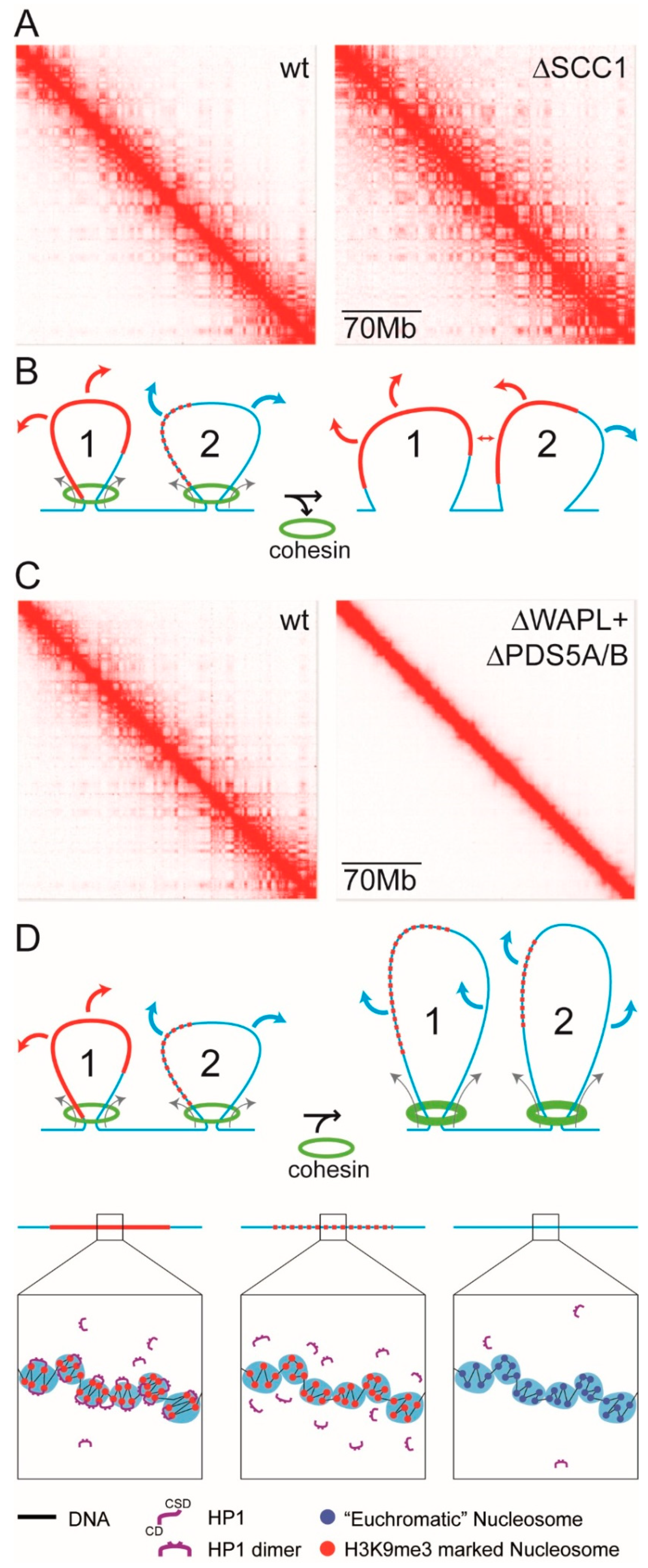
- The HTL term reduces the magnitude of χ and acts against phase separation. Accordingly, release of a domain/complex from the constraints of the chromatin fibre by severing linker DNA that connects the domain/complex to flanking euchromatin would remove the HTL term and enhance phase separation. Liquid Hi-C experiments have provided evidence that this is the case: chromatin fragmentation that releases domains/complexes from the chromatin fibre results in stronger compartmental segregation [190].
9. χN and the Order–Disorder Transition in Relation to Heterochromatin-Like and Pc-G Domains/Complexes
10. Conclusions and Perspectives
11. Coda
- The oligo-nucleosomal “clutch” is the “unit of incompatibility” of chromatin.
- When a domain/complex assembles from “clutches” consisting of “bridged” nucleosomes with zig-zag geometry it will have a tendency, specified by χ, for micro-phase separation from flanking euchromatin.
- A qualitative prediction of the degree of micro-phase separation (wavy vs. sharp interfaces) is specified by the magnitude of χN compared to χNODT for a given domain/complex.
- Segregation of micro-phase separated domains/complexes facilitates folding of the genome into the confines of the nucleus while retaining an environment that enables (regulation of) chromatin template-dependent processes.
Availability of Data and Materials
| Data | Original Paper | GEO Accession |
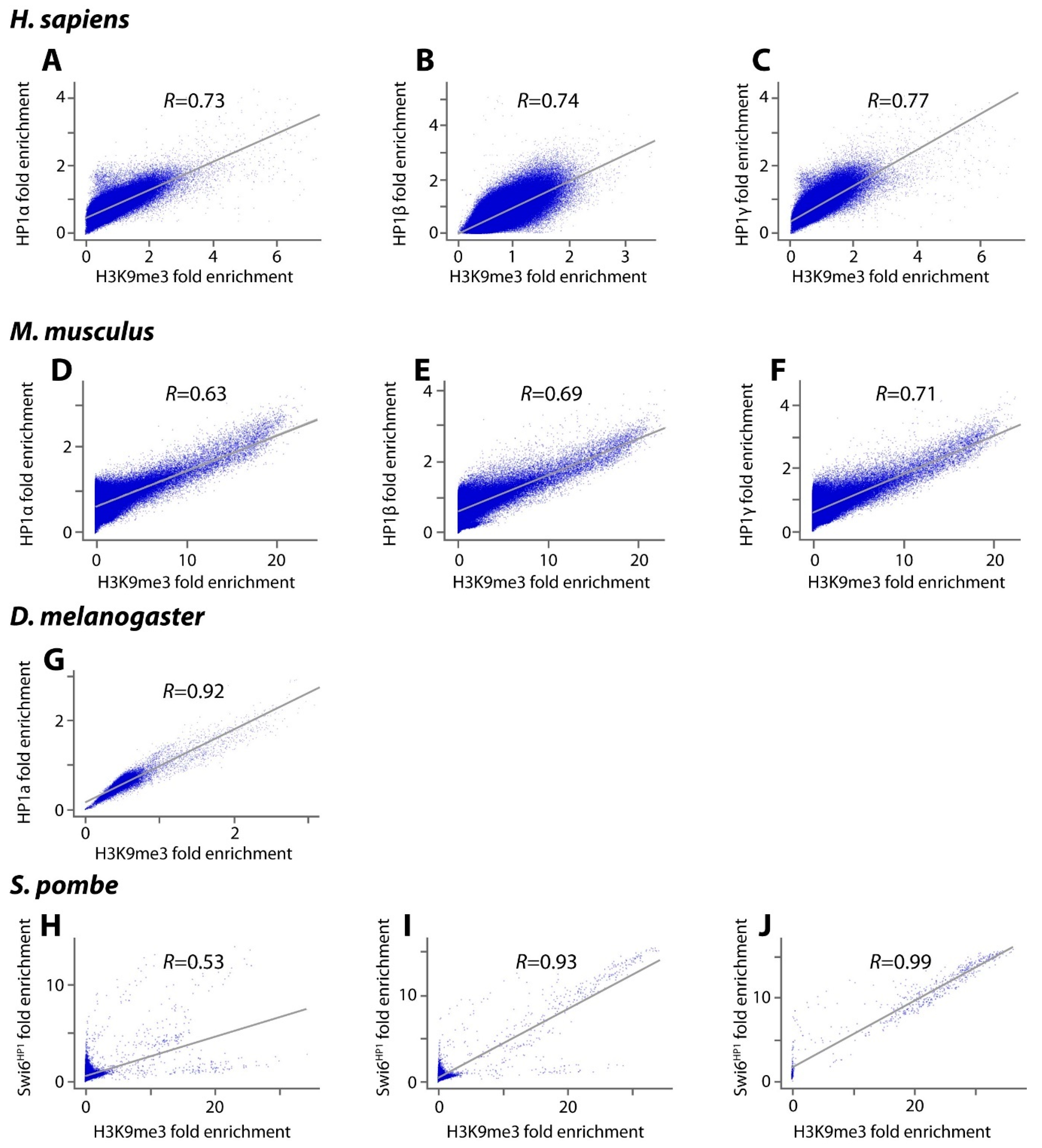
| HP1α (CBX5), HP1γ (CBX3), H3K9me3 in Human E1 ES cells | ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57-74, [59]. http://www.roadmapepigenomics.org/ [60] | GSE101135 GSE123229 GSE95868 GSE16256 |
| HP1α (CBX5), HP1β (CBX1), H3K9me3 in HEK293 cells | LeRoy, G.; Chepelev, I.; DiMaggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia B.A. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68, PMID: 22,897,906 [61]. Hattori, T.; Lai, D.; Dementieva, I.S.; Montaño, S.P.; Kurosawa, K.; Zheng, Y.; Akin, L.R.; Świst-Rosowska, K.M.; Grzybowski, A.T.; Koide, A.; Krajewski, K.; Strahl, B.D.; Kelleher, N.L.; Ruthenburg, A.J.; Koide, S. Antigen clasping by two antigen-binding sites of an exceptionally specific antibody for histone methylation. Proc Natl Acad Sci U S A. 2016, 113, 2092-2097, doi:10.1073/pnas.1522691113 [62]. | GSE39579 GSE66530 |
| HP1α (CBX5), HP1β (CBX1), HP1γ (CBX3), H3K9me3 in murine ES cells | Ostapcuk, V.; Mohn, F.; Carl, S.H.; Basters, A.; Hess, D.; Iesmantavicius, V.; Lampersberger, L.; Flemr, M.; Pandey, A.; Thomä, N.H.; Betschinger, J.; Bühler M. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature 2018, 557, 739-743, doi:10.1038/s41586-018-0153-8. [45]. | GSE97945 |
| HP1a and H3K9me3 in D. melanogaster | Klenov, M.S.; Lavrov, S.A.; Korbut, A.P.; Stolyarenko, A.D.; Yakushev, E.Y.; Reuter, M.; Pillai, R.S.; Gvozdev, V.A. Impact of nuclear Piwi elimination on chromatin state in Drosophila melanogaster ovaries. Nucleic Acids Res. 2014, 42, 6208-18. PMID: 24,782,529 [63]. | GSE56347 |
| Swi6HP1 and H3K9me3 in S. pombe | Iglesias, N.; Paulo, J.A.; Tatarakis, A.; Wang, X.; Edwards, A.L.; Bhanu, N.V.; Garcia, B.A.; Haas, W.; Gygi, S.P.; Moazed, D. Native Chromatin Proteomics Reveals a Role for Specific Nucleoporins in Heterochromatin Organization and Maintenance. Mol. Cell 2020, 77, 51-66.e8. PMID: 31,784,357 [64]. | GSE125935 |
Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wolffe, A.P. Chromatin: Structure and Function, 3rd ed.; Academic Press: London, UK, 2012. [Google Scholar]
- Lewis, E.B. The phenomenon of position effect. Adv. Genet. 1950, 3, 73–115. [Google Scholar] [PubMed]
- Baker, W.K. Position-effect variegation. In Advances in Genetics; Caspari, E.W., Ed.; Elsevier: Amsterdam, The Netherlands, 1968; Volume 14, pp. 133–169. [Google Scholar]
- Spofford, J.B. Position-effect variegation in Drosophila. In The Genetics and Biology of Drosophila; Ashburner, M., Novitski, E., Eds.; Academic Press: New York, NY, USA, 1976; Volume 1, pp. 955–1018. [Google Scholar]
- Elgin, S.C.; Reuter, G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J. The relation of the heterochromatic chromosome regions to the nucleic acids of the cell. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1956; Volume 21, pp. 307–328. [Google Scholar]
- Henikoff, S. Position-effect variegation and chromosome structure of a heat shock puff in Drosophila. Chromosoma 1981, 83, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Zhimulev, I.F.; Belyaeva, E.S.; Fomina, O.V.; Protopopov, M.O.; Bolshakov, V.N. Cytogenetic and molecular aspects of position effect variegation in Drosophila melanogaster. Chromosoma 1986, 94, 492–504. [Google Scholar] [CrossRef]
- Weiler, K.S.; Wakimoto, B.T. Heterochromatin and gene expression in Drosophila. Annu. Rev. Genet. 1995, 29, 577–605. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, T.; Yonezawa, M.; Lein, S.; Heidrich, K.; Kubicek, S.; Schäfer, C.; Phalke, S.; Walther, M.; Schmidt, A.; Jenuwein, T.; et al. Heterochromatin formation in Drosophila is initiated through active removal of H3K4 methylation by the LSD1 homolog SU (VAR) 3–3. Mol. Cell 2007, 26, 103–115. [Google Scholar] [CrossRef]
- Locke, J.; Kotarski, M.A.; Tartof, K.D. Dosage-dependent modifiers of position effect variegation in Drosophila and a mass action model that explains their effect. Genetics 1988, 120, 181–198. [Google Scholar]
- Wustmann, G.; Szidonya, J.; Taubert, H.; Reuter, G. The genetics of position-effect variegation modifying loci in Drosophila melanogaster. Mol. Gen. Genet. 1989, 217, 520–527. [Google Scholar] [CrossRef]
- Hayashi, S.; Ruddell, A.; Sinclair, D.; Grigliatti, T. Chromosomal structure is altered by mutations that suppress or enhance position effect variegation. Chromosoma 1990, 99, 391–400. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef] [PubMed]
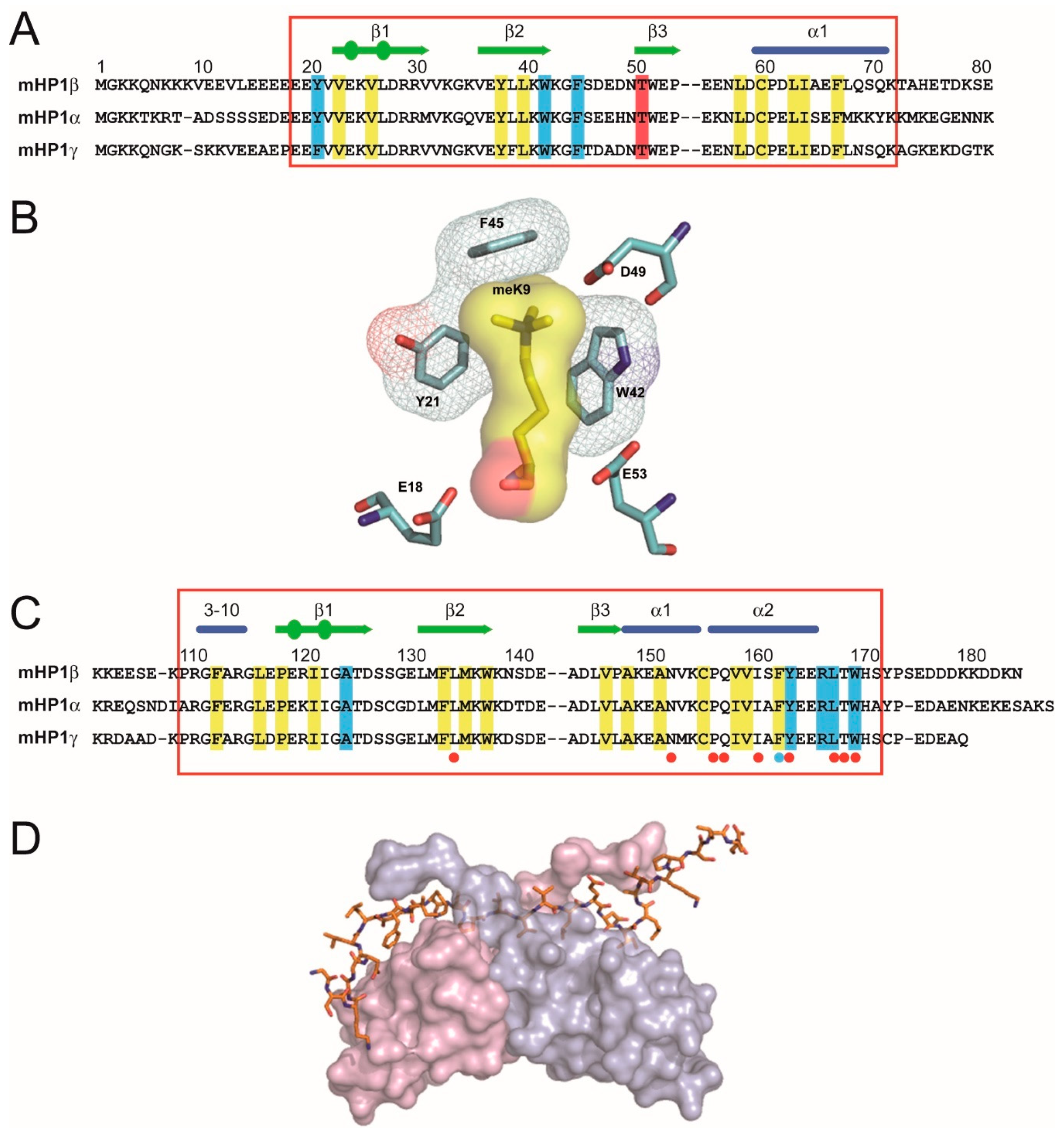
- Wang, G.; Ma, A.; Chow, C.M.; Horsley, D.; Brown, N.R.; Cowell, I.G.; Singh, P.B. Conservation of heterochromatin protein 1 function. Mol. Cell. Biol. 2000, 20, 6970–6983. [Google Scholar] [CrossRef] [PubMed]
- Elgin, S.C.; Grewal, S.I. Heterochromatin: Silence is golden. Curr. Biol. 2003, 13, R895–R898. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.P.; Sugiyama, T.; Chen, E.S.; Chen, X.; FitzGerald, P.C.; Grewal, S.I. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 2005, 37, 809–819. [Google Scholar] [CrossRef]
- Kellum, R.; Alberts, B.M. Heterochromatin protein 1 is required for correct chromosome segregation in Drosophila embryos. J. Cell Sci. 1995, 108, 1419–1431. [Google Scholar]
- Schotta, G.; Ebert, A.; Krauss, V.; Fischer, A.; Hoffmann, J.; Rea, S.; Jenuwein, T.; Dorn, R.; Reuter, G. Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002, 21, 1121–1131. [Google Scholar] [CrossRef]
- Fanti, L.; Giovinazzo, G.; Berloco, M.; Pimpinelli, S. The heterochromatin protein 1 prevents telomere fusions in Drosophila. Mol. Cell 1998, 2, 527–538. [Google Scholar] [CrossRef]
- Cowell, I.G.; Aucott, R.; Mahadevaiah, S.K.; Burgoyne, P.S.; Huskisson, N.; Bongiorni, S.; Prantera, G.; Fanti, L.; Pimpinelli, S.; Wu, R.; et al. Heterochromatin, HP1 and methylation at lysine 9 of histone H3 in animals. Chromosoma 2002, 111, 22–36. [Google Scholar] [CrossRef]
- Peng, J.C.; Karpen, G.H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef]
- García-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Sanyal, S.; Ghosh, A.; Bhar, K.; Das, C.; Siddhanta, A. Phosphatidylinositol-4-phosphate 5-kinase 1α modulates ribosomal RNA gene silencing through its interaction with histone H3 lysine 9 trimethylation and heterochromatin protein HP1-α. J. Biol. Chem. 2015, 290, 20893–20903. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Li, G.; Sikorski, T.W.; Buratowski, S.; Woodcock, C.L.; Moazed, D. Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol. Cell 2009, 35, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, R.F.; Feng, D.F.; Tsang, S.; Cho, G.; Little, E. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science 1996, 271, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Houseman, A.; Ivanov, A.R.; Wolf, D.A. Comparative proteomic and transcriptomic profiling of the fission yeast Schizosaccharomyces pombe. Mol. Syst. Biol. 2007, 3, 79. [Google Scholar] [CrossRef]
- Hiragami-Hamada, K.; Soeroes, S.; Nikolov, M.; Wilkins, B.; Kreuz, S.; Chen, C.; De La Rosa-Velázquez, I.A.; Zenn, H.M.; Kost, N.; Pohl, W.; et al. Dynamic and flexible H3K9me3 bridging via HP1β dimerization establishes a plastic state of condensed chromatin. Nat. Commun. 2016, 7, 11310. [Google Scholar] [CrossRef]
- Singh, P.B.; Miller, J.R.; Pearce, J.; Kothary, R.; Burton, R.D.; Paro, R.; James, T.C.; Gaunt, S.J. A sequence motif found in a Drosophila heterochromatin protein is conserved in animals and plants. Nucleic Acids Res. 1991, 19, 789–794. [Google Scholar] [CrossRef]
- Aucott, R.; Bullwinkel, J.; Yu, Y.; Shi, W.; Billur, M.; Brown, J.P.; Menzel, U.; Kioussis, D.; Wang, G.; Reisert, I.; et al. HP1-beta is required for development of the cerebral neocortex and neuromuscular junctions. J. Cell Biol. 2008, 183, 597–606. [Google Scholar] [CrossRef]
- Aasland, R.; Stewart, A.F. The chromo shadow domain, a second chromo domain in heterochromatin-binding protein 1, HP1. Nucleic Acids Res. 1995, 23, 3168–3173. [Google Scholar] [CrossRef]
- Epstein, H.; James, T.C.; Singh, P.B. Cloning and expression of Drosophila HP1 homologs from a mealybug Planococcus citri. J. Cell Sci. 1992, 101, 463–474. [Google Scholar]
- Ball, L.J.; Murzina, N.V.; Broadhurst, R.W.; Raine, A.R.; Archer, F.J.; Stott, F.J.; Murzin, A.G.; Singh, P.B.; Domaille, P.J.; Laue, E.D. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1. EMBO J. 1997, 16, 2473–2481. [Google Scholar] [CrossRef]
- Nielsen, P.R.; Nietlispach, D.; Mott, H.R.; Callaghan, J.; Bannister, A.; Kouzarides, T.; Murzin, A.G.; Murzina, N.V.; Laue, E.D. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 2002, 416, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cation-π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 2013, 113, 2100–2138. [Google Scholar] [CrossRef] [PubMed]
- Thiru, A.; Nietlispach, D.; Okuwaki, M.; Lyon, D.; Nielsen, P.R.; Hirshberg, M.; Verreault, A.; Murzina, N.V.; Laue, E.D. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004, 23, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Smothers, J.F.; Henikoff, S. The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol. Cell. Biol. 2001, 21, 2555–2569. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Billur, M.; Bartunik, H.D.; Singh, P.B. The essential function of HP1 beta: A case of the tail wagging the dog? Trends Biochem. Sci. 2010, 35, 115–123. [Google Scholar] [CrossRef]
- Singh, P.B. Molecular mechanisms of cellular determination: Their relation to chromatin structure and parental imprinting. J. Cell Sci. 1994, 107, 2653–2668. [Google Scholar]
- Jones, D.O.; Cowell, I.G.; Singh, P.B. Mammalian chromodomain proteins: Their role in genome organisation and expression. Bioessays 2000, 22, 124–137. [Google Scholar] [CrossRef]
- Singh, P.B.; Newman, A.G. On the relations of phase separation and Hi-C maps to epigenetics. R. Soc. Open Sci. 2020, 7, 191976. [Google Scholar] [CrossRef]
- Li, Y.; Kirschmann, D.A.; Wallrath, L.L. Does heterochromatin protein 1 always follow code? Proc. Natl. Acad. Sci. USA 2002, 99, 16462–16469. [Google Scholar] [CrossRef]
- Ostapcuk, V.; Mohn, F.; Carl, S.H.; Basters, A.; Hess, D.; Iesmantavicius, V.; Lampersberger, L.; Flemr, M.; Pandey, A.; Thomä, N.H.; et al. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature 2018, 557, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Cryderman, D.E.; Grade, S.K.; Li, Y.; Fanti, L.; Pimpinelli, S.; Wallrath, L.L. Role of Drosophila HP1 in euchromatic gene expression. Dev. Dyn. 2005, 232, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Perrini, B.; Piacentini, L.; Fanti, L.; Altieri, F.; Chichiarelli, S.; Berloco, M.; Turano, C.; Ferraro, A.; Pimpinelli, S. HP1 controls telomere capping, telomere elongation, and telomere silencing by two different mechanisms in Drosophila. Mol. Cell 2004, 15, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.L.; Oulad-Abdelghani, M.; Ortiz, J.A.; Remboutsika, E.; Chambon, P.; Losson, R. Heterochromatin formation in mammalian cells:interaction between histones and HP1 proteins. Mol. Cell 2001, 7, 729–739. [Google Scholar] [CrossRef]
- Lavigne, M.; Eskeland, R.; Azebi, S.; Saint-André, V.; Jang, S.M.; Batsché, E.; Fan, H.Y.; Kingston, R.E.; Imhof, A.; Muchardt, C. Interaction of HP1 and Brg1/Brm with the globular domain of histone H3 is required for HP1-mediated repression. PLoS Genet. 2009, 5, e1000769. [Google Scholar] [CrossRef] [PubMed]
- Richart, A.N.; Brunner, C.I.; Stott, K.; Murzina, N.V.; Thomas, J.O. Characterization of chromoshadow domain-mediated binding of heterochromatin protein 1α (HP1α) to histone H3. J. Biol. Chem. 2012, 287, 18730–18737. [Google Scholar] [CrossRef]
- Daujat, S.; Zeissler, U.; Waldemann, T.; Happel, N.; Schneider, R. HP1 binds Specifically to Lys26-methylated Histone 1.4, whereas Simultaneous Ser27 Phosphorylation Blocks HP1 binding. J. Biol. Chem. 2005, 280, 38090–38095. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Peters, A.H.; Quivy, J.-P.; Roche, D.; Taddei, A.; Lachner, M.; Jenuwein, T.; Almouzni, G. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 2002, 30, 329. [Google Scholar] [CrossRef]
- Muchardt, C.; Guillemé, M.; Seeler, J.-S.; Trouche, D.; Dejean, A.; Yaniv, M. Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1α. EMBO Rep. 2002, 3, 975–981. [Google Scholar] [CrossRef]
- Meehan, R.R.; Kao, C.-F.; Pennings, S. HP1 binding to native chromatin in vitro is determined by the hinge region and not by the chromodomain. EMBO J. 2003, 22, 3164–3174. [Google Scholar] [CrossRef]
- Sanulli, S.; Trnka, M.J.; Dharmarajan, V.; Tibble, R.W.; Pascal, B.D.; Burlingame, A.L.; Griffin, P.R.; Gross, J.D.; Narlikar, G.J. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Cheutin, T.; McNairn, A.J.; Jenuwein, T.; Gilbert, D.M.; Singh, P.B.; Misteli, T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 2003, 299, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Schmiedeberg, L.; Weisshart, K.; Diekmann, S.; Meyer zu Hoerste, G.; Hemmerich, P. High-and low mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell 2004, 15, 2819–2833. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, A.L.; Allshire, R.C. Kinetochore and heterochromatin domains of the fission yeast centromere. Chromosome Res. 2004, 12, 521–534. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Project. Available online: http://www.roadmapepigenomics.org/ (accessed on 15 March 2020).
- LeRoy, G.; Chepelev, I.; DiMaggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia, B.A. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68. [Google Scholar] [CrossRef]
- Hattori, T.; Lai, D.; Dementieva, I.S.; Montaño, S.P.; Kurosawa, K.; Zheng, Y.; Akin, L.R.; Świst-Rosowska, K.M.; Grzybowski, A.T.; Koide, A.; et al. Antigen clasping by two antigen-binding sites of an exceptionally specific antibody for histone methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 2092–2097. [Google Scholar] [CrossRef]
- Klenov, M.S.; Lavrov, S.A.; Korbut, A.P.; Stolyarenko, A.D.; Yakushev, E.Y.; Reuter, M.; Pillai, R.S.; Gvozdev, V.A. Impact of nuclear Piwi elimination on chromatin state in Drosophila melanogaster ovaries. Nucleic Acids Res. 2014, 42, 6208–6218. [Google Scholar] [CrossRef]
- Iglesias, N.; Paulo, J.A.; Tatarakis, A.; Wang, X.; Edwards, A.L.; Bhanu, N.V.; Garcia, B.A.; Haas, W.; Gygi, S.P.; Moazed, D. Native Chromatin Proteomics Reveals a Role for Specific Nucleoporins in Heterochromatin Organization and Maintenance. Mol. Cell 2020, 77, 51–66.e8. [Google Scholar] [CrossRef]
- Singh, P.B. Heterochromatin and the molecular mechanisms of ‘parent-of-origin’ effects in animals. J. Biosci. 2016, 41, 759–786. [Google Scholar] [CrossRef]
- Horsley, D.; Hutchings, A.; Butcher, G.W.; Singh, P.B. M32, a murine homologue of Drosophila heterochromatin protein 1 (HP1), localises to euchromatin within interphase nuclei and is largely excluded from constitutive heterochromatin. Cytogenet Cell Genet. 1996, 73, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.J.; Guelen, L.; de Wit, E.; Peric-Hupkes, D.; Lodén, M.; Talhout, W.; Feenstra, M.; Abbas, B.; Classen, A.K.; van Steensel, B. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 2006, 16, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef]
- Hediger, F.; Gasser, S.M. Heterochromatin protein 1: Don’t judge the book by its cover! Curr. Opin. Genet. Dev. 2006, 16, 143–150. [Google Scholar] [CrossRef]
- Kwon, S.H.; Workman, J.L. The heterochromatin protein 1 (HP1) family: Put away a bias toward HP1. Mol. Cell 2008, 26, 217–227. [Google Scholar]
- Piacentini, L.; Fanti, L.; Berloco, M.; Perrini, B.; Pimpinelli, S. Heterochromatin protein 1 (HP1) is associated with induced gene expression in Drosophila euchromatin. J. Cell Biol. 2003, 161, 707–714. [Google Scholar] [CrossRef]
- Smallwood, A.; Hon, G.C.; Jin, F.; Henry, R.E.; Espinosa, J.M.; Ren, B. CBX3 regulates efficient RNA processing genome-wide. Genome Res. 2012, 22, 1426–1436. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Yearim, A.; Gelfman, S.; Shayevitch, R.; Melcer, S.; Glaich, O.; Mallm, J.P.; Nissim-Rafinia, M.; Cohen, A.H.; Rippe, K.; Meshorer, E.; et al. HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 2015, 10, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Dinant, C.; Luijsterburg, M.S. The emerging role of HP1 in the DNA damage response. Mol. Cell. Biol. 2009, 29, 6335–6340. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Bateson, W. Mendel’s Principles of Heredity; Cambridge University Press: Cambridge, UK, 1909. [Google Scholar]
- Mendel, G. Versuche uber Pflanzen-Hybriden. Verh. Naturforsch. Ver. Brunn 1866, 4, 3–47, English translation in J. R. Hortic. Soc. 1901, 26, 1–32. [Google Scholar]
- Metz, C.W. Chromosome behavior, inheritance and sex determination in Sciara. Am. Nat. 1938, 72, 485–520. [Google Scholar] [CrossRef]
- Brown, S.W.; Nur, U. Heterochromatic chromosomes in the coccids. Science 1964, 145, 130–136. [Google Scholar] [CrossRef]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Surani, M.A.; Barton, S.C.; Norris, M.L. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 1984, 308, 548–550. [Google Scholar] [CrossRef]
- Henckel, A.; Arnaud, P. Genome-wide identification of new imprinted genes. Brief. Funct. Genomics 2010, 9, 304–314. [Google Scholar] [CrossRef]
- Williamson, C.M.; Blake, A.; Thomas, S.; Beechey, C.V.; Hancock, J.; Cattanach, B.M.; Peters, J.; MRC Harwell; Oxfordshire. World Wide Web Site-Mouse Imprinting Data and References. Available online: http://www.har.mrc.ac.uk/research/genomic_imprinting (accessed on 16 July 2020).
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Tomizawa, S.; Kobayashi, H.; Watanabe, T.; Andrews, S.; Hata, K.; Kelsey, G.; Sasaki, H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 2011, 138, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Suda, C.; Abe, T.; Kohara, Y.; Ikemura, T.; Sasaki, H. Bisulfite sequencing and dinucleotide content analysis of 15 imprinted mouse differentially methylated regions (DMRs): Paternally methylated DMRs contain less CpGs than maternally methylated DMRs. Cytogenet Genome Res. 2006, 113, 130–137. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Duan, J.; Gao, X.; Zhu, W.; Lu, X.; Yang, L.; Zhang, J.; Li, G.; Ci, W.; et al. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef]
- Hanna, C.W.; Kelsey, G. The specification of imprints in mammals. Hered. 2014, 113, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Sakurai, T.; Imai, M.; Takahashi, N.; Fukuda, A.; Yayoi, O.; Sato, S.; Nakabayashi, K.; Hata, K.; Sotomaru, Y.; et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012, 8, e1002440. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.R.; Veselovska, L.; Kelsey, G. Establishment and functions of DNA methylation in the germline. Epigenomics 2016, 8, 1399–1413. [Google Scholar] [CrossRef]
- Sendžikaitė, G.; Kelsey, G. The role and mechanisms of DNA methylation in the oocyte. Essays Biochem. 2019, 63, 691–705. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, 339–344. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Leseva, M.; Knowles, B.B.; Messerschmidt, D.M.; Solter, D. Erase-Maintain-Establish: Natural Reprogramming of the Mammalian Epigenome. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 155–163. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef]
- Takahashi, N.; Coluccio, A.; Thorball, C.W.; Planet, E.; Shi, H.; Offner, S.; Turelli, P.; Imbeault, M.; Ferguson-Smith, A.C.; Trono, D. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 2019, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Anvar, Z.; Cammisa, M.; Riso, V.; Baglivo, I.; Kukreja, H.; Sparago, A.; Girardot, M.; Lad, S.; De Feis, I.; Cerrato, F.; et al. ZFP57 recognizes multiple and closely spaced sequence motif variants to maintain repressive epigenetic marks in mouse embryonic stem cells. Nucleic Acids Res. 2016, 44, 1118–1132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Toh, H.; Sasaki, H.; Zhang, X.; Cheng, X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012, 26, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; de Vries, W.; Ito, M.; Solter, D.; Ferguson-Smith, A.; Knowles, B.B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012, 335, 1499–1502. [Google Scholar] [CrossRef]
- Cho, S.; Park, J.S.; Kwon, S.; Kang, Y.K. Dynamics of Setdb1 expression in early mouse development. Gene Expr. Patterns. 2012, 12, 213–218. [Google Scholar] [CrossRef]
- Kim, J.; Zhao, H.; Dan, J.; Kim, S.; Hardikar, S.; Hollowell, D.; Lin, K.; Lu, Y.; Takata, Y.; Shen, J.; et al. Maternal Setdb1 Is Required for Meiotic Progression and Preimplantation Development in Mouse. PLoS Genet. 2016, 12, e1005970. [Google Scholar] [CrossRef]
- Leung, D.; Du, T.; Wagner, U.; Xie, W.; Lee, A.Y.; Goyal, P.; Li, Y.; Szulwach, K.E.; Jin, P.; Lorincz, M.C.; et al. Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc. Natl. Acad. Sci. USA 2014, 111, 6690–6695. [Google Scholar] [CrossRef]
- Arney, K.L.; Bao, S.; Bannister, A.J.; Kouzarides, T.; Surani, M.A. Histone methylation defines epigenetic asymmetry in the mouse zygote. Int. J. Dev. Biol. 2002, 46, 317–320. [Google Scholar]
- Probst, A.V.; Santos, F.; Reik, W.; Almouzni, G.; Dean, W. Structural differences in centromeric heterochromatin are spatially reconciled on fertilisation in the mouse zygote. Chromosoma 2007, 116, 403–415. [Google Scholar] [CrossRef]
- Zuo, X.; Sheng, J.; Lau, H.T.; McDonald, C.M.; Andrade, M.; Cullen, D.E.; Bell, F.T.; Iacovino, M.; Kyba, M.; Xu, G.; et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 2012, 287, 2107–2118. [Google Scholar] [CrossRef]
- Cirio, M.C.; Ratnam, S.; Ding, F.; Reinhart, B.; Navara, C.; Chaillet, J.R. Preimplantation expression of the somatic form of Dnmt1 suggests a role in the inheritance of genomic imprints. BMC Dev. Biol. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kawamura, Y.; Uchijima, Y.; Amamo, T.; Kobayashi, H.; Asano, T.; Kurihara, H. Maintenance of genomic methylation patterns during preimplantation development requires the somatic form of DNA methyltransferase 1. Dev. Biol. 2008, 313, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, R.; Chiba, H.; Kaneda, M.; Tajima, S.; Li, E.; Jaenisch, R.; Sasaki, H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008, 22, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Rodriguez-Terrones, D.; Burton, A.; Torres-Padilla, M.E. SUV4-20 activity in the preimplantation mouse embryo controls timely replication. Genes Dev. 2016, 30, 2513–2526. [Google Scholar] [CrossRef]
- Wongtawan, T.; Taylor, J.E.; Lawson, K.A.; Wilmut, I.; Pennings, S. Histone H4K20me3 and HP1α are late heterochromatin markers in development, but present in undifferentiated embryonic stem cells. J. Cell Sci. 2011, 124, 1878–1890. [Google Scholar] [CrossRef]
- Smith, Z.D.; Chan, M.M.; Humm, K.C.; Karnik, R.; Mekhoubad, S.; Regev, A.; Eggan, K.; Meissner, A. DNA methylation dynamics of the human preimplantation embryo. Nature 2014, 511, 611–615. [Google Scholar] [CrossRef]
- Strogantsev, R.; Krueger, F.; Yamazawa, K.; Shi, H.; Gould, P.; Goldman-Roberts, M.; McEwen, K.; Sun, B.; Pedersen, R.; Ferguson-Smith, A.C. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015, 16, 112. [Google Scholar] [CrossRef]
- Pannetier, M.; Julien, E.; Schotta, G.; Tardat, M.; Sardet, C.; Jenuwein, T.; Feil, R. PR-SET7 and SUV4-20H regulate H4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep. 2008, 9, 998–1005. [Google Scholar] [CrossRef]
- Regha, K.; Sloane, M.A.; Huang, R.; Pauler, F.M.; Warczok, K.E.; Melikant, B.; Radolf, M.; Martens, J.H.; Schotta, G.; Jenuwein, T.; et al. Active and repressive chromatin are interspersed without spreading in an imprinted gene cluster in the mammalian genome. Mol. Cell 2007, 27, 353–366. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Singh, P.B.; Shloma, V.V.; Belyakin, S.N. Maternal regulation of chromosomal imprinting in animals. Chromosoma 2019, 128, 69–80. [Google Scholar] [CrossRef]
- Zeng, T.B.; Han, L.; Pierce, N.; Pfeifer, G.P.; Szabó, P.E. EHMT2 and SETDB1 protect the maternal pronucleus from 5mC oxidation. Proc. Natl. Acad. Sci. USA 2019, 116, 10834–10841. [Google Scholar] [CrossRef]
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, L. The Triumph of the Embryo; Oxford University Press: Oxford, UK, 1991; ISBN 0-19-854243-7. [Google Scholar]
- Nicetto, D.; Donahue, G.; Jain, T.; Peng, T.; Sidoli, S.; Sheng, L.; Montavon, T.; Becker, J.S.; Grindheim, J.M.; Blahnik, K.; et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 2019, 363, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Haeckel, E. Anthropogenie oder Entwurklungsgeschuchte des Menschen, 4th ed.; Engelmann: Leipzig, Germany, 1891. [Google Scholar]
- Hopwood, N. Haeckel’s Embryos: Images, Evolution and Fraud; The University of Chicago Press: Chicago, IL, USA, 2015; ISBN 978-0-226-04694-5. [Google Scholar]
- Sander, K. The evolution of patterning mechanisms: Gleanings from insect embryogenesis and spermatogenesis. In Development and Evolution; Goodwin, B.C., Holder, N., Wylie, C.G., Eds.; Cambridge University Press: Cambridge, UK, 1983; pp. 137–159. [Google Scholar]
- Richardson, M.K. Heterochrony and the phylotypic period. Dev. Biol. 1995, 172, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.K. A phylotypic stage for all animals? Dev. Cell 2012, 22, 903–904. [Google Scholar] [CrossRef] [PubMed]
- Duboule, D. Temporal colinearity and the phylotypic progression: A basis for the stability of a vertebrate Bauplan and the evolution of morphologies through heterochrony. Dev. Suppl. 1994, 135–142. [Google Scholar]
- Raff, R.A. Developmental mechanisms in the evolution of animal form: Origins and evolvability of body plans. In Early Life on Earth; Bengston, S., Ed.; Columbia: New York, NY, USA, 1994; pp. 489–500. [Google Scholar]
- Irie, N.; Kuratani, S. The developmental hourglass model: A predictor of the basic body plan? Development 2014, 141, 4649–4655. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat. Commun. 2011, 2, 248. [Google Scholar] [CrossRef]
- Slack, J.M.; Holland, P.W.; Graham, C.F. The zootype and the phylotypic stage. Nature 1993, 361, 490–492. [Google Scholar] [CrossRef]
- Young, T.; Deschamps, J. Hox, Cdx, and anteroposterior patterning in the mouse embryo. Curr. Top. Dev. Biol. 2009, 88, 235–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, X.; Gao, Y.; Yang, L.; Li, C.; Liu, W.; Chen, C.; Kou, X.; Zhao, Y.; Chen, J.; et al. Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat. Cell Biol. 2018, 20, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Elling, U.; Hopfgartner, B.; Jung, Y.L.; Murn, J.; Ninova, M.; Hubmann, M.; Badeaux, A.I.; Euong Ang, C.; Tenen, D.; et al. The histone chaperone CAF-1 safeguards somatic cell identity. Nature 2015, 528, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Borkent, M.; Bennett, B.D.; Lackford, B.; Bar-Nur, O.; Brumbaugh, J.; Wang, L.; Du, Y.; Fargo, D.C.; Apostolou, E.; Cheloufi, S.; et al. A serial shRNA screen for roadblocks to reprogramming identifies the protein modifier SUMO2. Stem Cell Rep. 2016, 6, 704–716. [Google Scholar] [CrossRef]
- Noordermeer, D.; Duboule, D. Chromatin looping and organization at developmentally regulated gene loci. Wiley Interdiscip Rev. Dev. Biol. 2013, 2, 615–630. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Botting, C.H.; Grimes, G.R.; Bickmore, W.A.; Eskeland, R. PRC1 and PRC2 are not required for targeting of H2A.Z to developmental genes in embryonic stem cells. PLoS ONE 2012, 7, e34848. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 2016, 63, 1066–1079. [Google Scholar] [CrossRef]
- Noordermeer, D.; Leleu, M.; Splinter, E.; Rougemont, J.; De Laat, W.; Duboule, D. The dynamic architecture of Hox gene clusters. Science 2011, 334, 222–225. [Google Scholar] [CrossRef]
- Neijts, R.; Simmini, S.; Giuliani, F.; van Rooijen, C.; Deschamps, J. Region-specific regulation of posterior axial elongation during vertebrate embryogenesis. Dev. Dyn. 2014, 243, 88–98. [Google Scholar] [CrossRef]
- Deschamps, J.; Duboule, D. Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes Dev. 2017, 31, 1406–1416. [Google Scholar] [CrossRef]
- Soshnikova, N.; Duboule, D. Epigenetic temporal control of mouse Hox genes in vivo. Science 2009, 324, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Vieux-Rochas, M.; Fabre, P.J.; Leleu, M.; Duboule, D.; Noordermeer, D. Clustering of mammalian Hox genes with other H3K27me3 targets within an active nuclear domain. Proc. Natl. Acad. Sci. USA 2015, 112, 4672–4677. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.C.; Nelson, C.E.; Morgan, B.A.; Tabin, C. Hox genes and the evolution of vertebrate axial morphology. Development 1995, 121, 333–346. [Google Scholar] [PubMed]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics Consortium Roadmap; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317. [Google Scholar] [CrossRef] [PubMed]
- Molitor, J.; Mallm, J.-P.; Rippe, K.; Erdel, F. Retrieving chromatin patterns fromdeep sequencing data using correlation functions. Biophys. J. 2017, 112, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Matsen, M.W. Self-consistent Field Theory and Its Applications. Soft Matter 2007, 1, 87–178. [Google Scholar] [CrossRef]
- Riess, G. Micellization of block copolymers. Prog. Polym. Sci. 2003, 28, 1107–1170. [Google Scholar] [CrossRef]
- Bates, F.S.; Fredrickson, G.H. Block copolymer thermodynamics: Theory and experiment. Annu. Rev. Phys. Chem. 1990, 41, 525–557. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of High Polymer Solutions. J. Chem. Phys. 1941, 9, 660. [Google Scholar] [CrossRef]
- Huggins, M.L. Solutions of Long Chain Compounds. J. Chem. Phys. 1941, 9, 440. [Google Scholar] [CrossRef]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2017, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Mirny, L.A.; Imakaev, M.; Abdennur, N. Two major mechanisms of chromosome organization. Curr. Opin Cell Biol. 2019, 58, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef]
- Vian, L.; Pękowska, A.; Rao, S.S.P.; Kieffer-Kwon, K.R.; Jung, S.; Baranello, L.; Huang, S.C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The energetics and physiological impact of cohesin extrusion. Cell 2018, 173, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e24. [Google Scholar] [CrossRef]
- Parmar, J.J.; Woringer, M.; Zimmer, C. How the genome Folds: The Biophysics of Four-Dimensional Chromatic Organisation. Ann. Rev. Biophys. 2019, 48, 231–253. [Google Scholar] [CrossRef]
- Wutz, G.; Várnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef]
- Zhou, L.; Liang, C.; Chen, Q.; Zhang, Z.; Zhang, B.; Yan, H.; Qi, F.; Zhang, M.; Yi, Q.; Guan, Y.; et al. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 2010, 143, 737–749. [Google Scholar] [CrossRef]
- Ouyang, Z.; Zheng, G.; Tomchick, D.R.; Luo, X.; Yu, H. Structural Basis and IP6 Requirement for Pds5-Dependent Cohesin Dynamics. Mol. Cell 2016, 62, 248–259. [Google Scholar] [CrossRef]
- Buheitel, J.; Stemmann, O. Prophase pathway-dependent removal of cohesin from human chromosomes requires opening of the Smc3-Scc1 gate. EMBO J. 2013, 32, 666–676. [Google Scholar] [CrossRef]
- Eichinger, C.S.; Kurze, A.; Oliveira, R.A.; Nasmyth, K. Disengaging the Smc3/kleisin interface releases cohesin from Drosophila chromosomes during interphase and mitosis. EMBO J. 2013, 32, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Gillespie, P.J.; Hirano, T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr. Biol. 2006, 16, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Haarhuis, J.H.; Elbatsh, A.M.; van den Broek, B.; Camps, D.; Erkan, H.; Jalink, K.; Medema, R.H.; Rowland, B.D. WAPL-mediated removal of cohesin protects against segregation errors and aneuploidy. Curr Biol. 2013, 23, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Kueng, S.; Hegemann, B.; Peters, B.H.; Lipp, J.J.; Schleiffer, A.; Mechtler, K.; Peters, J.M. Wapl controls the dynamic association of cohesin with chromatin. Cell 2006, 127, 955–967. [Google Scholar] [CrossRef]
- Folco, H.D.; McCue, A.; Balachandran, V.; Grewal, S.I.S. Cohesin Impedes Heterochromatin Assembly in Fission Yeast Cells Lacking Pds5. Genetics 2019, 213, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Rudra, S.; Skibbens, R.V. Cohesin codes—Interpreting chromatin architecture and the many facets of cohesin function. J. Cell Sci. 2013, 126, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Tapia-Alveal, C.; Jabado, O.J.; Germain, D.; O’Connell, M.J. An acetyltransferase-independent function of Eso1 regulates centromere cohesion. Mol. Biol. Cell 2016, 27, 4002–4010. [Google Scholar] [CrossRef]
- Hamley, I.W. The Physics of Block Copolymers; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Bates, F.S.; Frederickson, G.H. Block Copolymers—Designer Soft Materials. Physics Today 1999, 52, 32. [Google Scholar] [CrossRef]
- Bates, F.S. Polymer-polymer phase behavior. Science 1991, 251, 898–905. [Google Scholar] [CrossRef]
- Chua, E.Y.; Vogirala, V.K.; Inian, O.; Wong, A.S.; Nordenskiöld, L.; Plitzko, J.M.; Danev, R.; Sandin, S. 3.9 Å structure of the nucleosome core particle determined by phase-plate cryo-EM. Nucleic Acids Res. 2016, 44, 8013–8019. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Imai, R.; Tamura, S.; Nozaki, T. Chromatin as dynamic 10-nm fibers. Chromosoma 2014, 123, 225–237. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Grosberg, A.; Khokhlov, A.R. Giant Molecules: Here, There, and Everywhere; Academic Press: New York, NY, USA, 1997; ISBN1 0123041309. ISBN2 9780123041302. [Google Scholar]
- Azzaz, A.M.; Vitalini, M.W.; Thomas, A.S.; Price, J.P.; Blacketer, M.J.; Cryderman, D.E.; Zirbel, L.N.; Woodcock, C.L.; Elcock, A.H.; Wallrath, L.L.; et al. Human heterochromatin protein 1α promotes nucleosome associations that drive chromatin condensation. J. Biol. Chem. 2014, 289, 6850–6861. [Google Scholar] [CrossRef] [PubMed]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.; Wolf, M.; Kurumizaka, H. Structural basis of heterochromatin formation by human HP1. Mol. Cell 2018, 69, 385–397. [Google Scholar] [CrossRef]
- Belaghzal, H.; Borrman, T.; Stephens, A.D.; Lafontaine, D.L.; Venev, S.V.; Weng, Z.; Marko, J.F.; Dekker, J. Compartment-dependent chromatin interaction dynamics revealed by liquid chromatin Hi-C. bioRxiv 2019, 704957. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef]
- Nicodemi, M.; Pombo, A. Models of chromosome structure. Curr. Opin. Cell Biol. 2014, 28, 90–95. [Google Scholar] [CrossRef]
- MacPherson, Q.; Beltran, B.; Spakowitz, A.J. Bottom-up modeling of chromatin segregation due to epigenetic modifications. Proc. Natl. Acad. Sci. USA 2018, 115, 12739–12744. [Google Scholar] [CrossRef]
- Grau, D.J.; Chapman, B.A.; Garlick, J.D.; Borowsky, M.; Francis, N.J.; Kingston, R.E. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Genes Dev. 2011, 25, 2210–2221. [Google Scholar] [CrossRef]
- Kim, J.; Kingston, R.E. The CBX family of proteins in transcriptional repression and memory. J. Biosci. 2020, 45, 16. [Google Scholar] [CrossRef]
- Krietenstein, N.; Rando, O.J. Mesoscale organization of the chromatin fiber. Curr. Opin Genet. Dev. 2020, 61, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lim, H.H.; Shi, J.; Tamura, S.; Maeshima, K.; Surana, U.; Gan, L. Budding yeast chromatin is dispersed in a crowded nucleoplasm in vivo. Mol. Biol. Cell 2016, 27, 3357–3368. [Google Scholar] [CrossRef]
- Dubochet, J.; Adrian, M.; Schultz, P.; Oudet, P. Cryo-electron microscopy of vitrified SV40 minichromosomes: The liquid drop model. EMBO J. 1986, 5, 519–528. [Google Scholar] [CrossRef]
- Eltsov, M.; Maclellan, K.M.; Maeshima, K.; Frangakis, A.S.; Dubochet, J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl. Acad. Sci. USA 2008, 105, 19732–19737. [Google Scholar] [CrossRef]
- Risca, V.I.; Denny, S.K.; Straight, A.F.; Greenleaf, W.J. Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature 2017, 541, 237. [Google Scholar] [CrossRef]
- Rydberg, B.; Holley, W.R.; Mian, I.S.; Chatterjee, A. Chromatin conformation in living cells: Support for a zig-zag model of the 30 nm chromatin fiber. J. Mol. Biol. 1998, 284, 71–84. [Google Scholar] [CrossRef]
- Grigoryev, S.A.; Arya, G.; Correll, S.; Woodcock, C.L.; Schlick, T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. USA 2009, 106, 13317–13322. [Google Scholar] [CrossRef]
- Schalch, T.; Duda, S.; Sargent, D.F.; Richmond, T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fiber. Nature 2005, 436, 138–141. [Google Scholar] [CrossRef]
- Ekundayo, B.; Richmond, T.J.; Schalch, T. Capturing Structural Heterogeneity in Chromatin Fibers. J. Mol. Biol. 2017, 429, 3031–3042. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Chen, P.; Sun, D.; Wang, M.; Dong, L.; Liang, D.; Xu, R.M.; Zhu, P.; Li, G. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 2014, 344, 376–380. [Google Scholar] [CrossRef]
- Lohr, D.; Van Holde, K.E. Organization of spacer DNA in chromatin. Proc. Natl. Acad. Sci. USA 1979, 76, 6326–6330. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, D.; Zhurkin, V.B. Topological polymorphism of the two-start chromatin fiber. Biophys. J. 2015, 108, 2591–2600. [Google Scholar] [CrossRef]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.G.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484.e21. [Google Scholar] [CrossRef] [PubMed]
- Brasher, S.V.; Smith, B.O.; Fogh, R.H.; Nietlispach, D.; Thiru, A.; Nielsen, P.R.; Broadhurst, R.W.; Ball, L.J.; Murzina, N.V.; Laue, E.D. The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer. EMBO J. 2000, 19, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, P.J.; Koslover, E.F.; Spakowitz, A.J. Thermodynamic model of heterochromatin formation through epigenetic regulation. J. Phys. Condens Matter 2015, 27, 064109. [Google Scholar] [CrossRef]
- Saksouk, N.; Simboeck, E.; Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 2015, 8, 3. [Google Scholar] [CrossRef]
- Teif, V.B.; Kepper, N.; Yserentant, K.; Wedemann, G.; Rippe, K. Affinity, stoichiometry and cooperativity of heterochromatin protein 1 (HP1) binding to nucleosomal arrays. J. Phys. Condens Matter 2015, 27, 064110. [Google Scholar] [CrossRef]
- Al-Sady, B.; Madhani, H.D.; Narlikar, G.J. Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Mol. Cell 2013, 51, 80–91. [Google Scholar] [CrossRef]
- Canzio, D.; Chang, E.Y.; Shankar, S.; Kuchenbecker, K.M.; Simon, M.D.; Madhani, H.D.; Narlikar, G.J.; Al-Sady, B. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol. Cell 2011, 41, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Reinberg, D.; Vales, L.D. Chromatin domains rich in inheritance. Science 2018, 361, 33–34. [Google Scholar] [CrossRef]
- Leibler, L. Theory of Microphase Separation in Block Copolymers. Macromolecules 1980, 13, 1602. [Google Scholar] [CrossRef]
- Edwards, S.F. The statistical mechanics of polymers with excluded volume. Proc. Phys. Soc. 1965, 85, 613–624. [Google Scholar] [CrossRef]
- MacPherson, Q.; Beltran, B.; Spakowitz, A.J. Chromatin Compaction Leads to a Preference for Peripheral Heterochromatin. Biophys. J. 2020, 118, 1479–1488. [Google Scholar] [CrossRef]
- Mau, S.; MacPherson, Q.; Spakowitz, A.J. Polymer Semiflexibility Induces Nonuniversal Phase Transitions in Diblock Copolymers. Phys. Rev. Lett. 2018, 120, 067802–067805. [Google Scholar] [CrossRef]
- Shin, Y.; Chang, Y.-C.; Lee, D.S.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid nuclear condensates mechanically sense and restructure the genome. Cell 2018, 175, 1481–1491.e13. [Google Scholar] [CrossRef]
- Plys, A.J.; Davis, C.P.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Tatavosian, R.; Kent, S.; Brown, K.; Yao, T.; Duc, H.N.; Huynh, T.N.; Zhen, C.Y.; Ma, B.; Wang, H.; Ren, X. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019, 294, 1451–1463. [Google Scholar] [CrossRef]
- Kaustov, L.; Ouyang, H.; Amaya, M.; Lemak, A.; Nady, N.; Duan, S.; Wasney, G.A.; Li, Z.; Vedadi, M.; Schapira, M.; et al. Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem. 2011, 286, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.M.; Shankaranarayana, G.D.; Noma, K.; Ayoub, N.; Cohen, A.; Grewal, S.I. Establishment and maintenance of a heterochromatin domain. Science 2002, 297, 2232–2237. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef]
- Helleboid, P.Y.; Heuse, l.M.; Duc, J.; Piot, C.; Thorball, C.W.; Coluccio, A.; Pontis, J.; Imbeault, M.; Turelli, P.; Aebersold, R.; et al. The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification. EMBO J. 2019, 38, e101220. [Google Scholar] [CrossRef]
- Yang, P.; Wang, Y.; Macfarlan, T.S. The Role of KRAB-ZFPs in Transposable Element Repression and Mammalian Evolution. Trends Genet. 2017, 33, 871–881. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Bestor, T.H.; Edwards, J.R.; Boulard, M. Notes on the role of dynamic DNA methylation in mammalian development. Proc. Natl. Acad. Sci. USA 2015, 112, 6796–6799. [Google Scholar] [CrossRef]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendergast, J.G.; Mjoseng, H.; et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013, 14, R25. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1, e00205. [Google Scholar] [CrossRef]
- Riising, E.M.; Comet, I.; Leblanc, B.; Wu, X.; Johansen, J.V.; Helin, K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 2014, 55, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, K.; Flyamer, I.M.; Thomson, J.P.; Mjoseng, H.K.; Shukla, R.; Williamson, I.; Grimes, G.R.; Illingworth, R.S.; Adams, I.R.; Pennings, S.; et al. DNA Methylation Directs Polycomb-Dependent 3D Genome Re-organization in Naive Pluripotency. Cell Rep. 2019, 29, 1974–1985.e6. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Hihara, S.; Eltsov, M. Chromatin structure: Does the 30-nm fiber exist in vivo? Curr. Opin. Cell Biol. 2010, 22, 291–297. [Google Scholar] [CrossRef]
- Maeshima, K.; Tamura, S.; Hansen, J.C.; Itoh, Y. Fluid-like chromatin: Toward understanding the real chromatin organization present in the cell. Curr. Opin. Cell Biol. 2020, 64, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983. [Google Scholar] [CrossRef]
- Wang, Z.; Patel, D.J. Combinatorial readout of dual histone modifications by paired chromatin-associated modules. J. Biol. Chem. 2011, 286, 18363–18368. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Amemiya, H.M.; Kundaje, A.; Boyle, A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019, 9, 9354. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Size, Mb | Cell Type|Components | Whole Genome | Heterochromatic Regions Excluded |
|---|---|---|---|---|
| H. sapiens | >1 | H1 ES cells (HP1α + HP1γ + H3K9me3) | 48 | 4 |
| 0.1–1 | H1 ES cells (HP1α + HP1γ + H3K9me3) | 345 | 159 | |
| 0.01–0.1 | H1 ES cells (HP1α + HP1γ + H3K9me3) | 19,550 | 18,853 | |
| >1 | 293T cells (HP1α + HP1β + H3K9me3) | 24 | 4 | |
| 0.1–1 | 293T cells (HP1α + HP1β + H3K9me3) | 1027 | 855 | |
| 0.01–0.1 | 293T cells (HP1α + HP1β + H3K9me3) | 33,754 | 32,292 | |
| M. musculus | >1 | ES cells (HP1α + HP1β + HP1γ + H3K9me3) | 0 | 0 |
| 0.1–1 | ES cells (HP1α + HP1β + HP1γ + H3K9me3) | 1059 | 622 | |
| 0.01–0.1 | ES cells (HP1α + HP1β + HP1γ + H3K9me3) | 12,675 | 10,227 | |
| D. melanogaster | >1 | Ovaries (HP1a + H3K9me3) | 7 | 0 |
| 0.1–1 | Ovaries (HP1a + H3K9me3) | 27 | 2 | |
| 0.01–0.1 | Ovaries (HP1a + H3K9me3) | 183 | 161 | |
| S. pombe | >1 | (Swi6HP1 + H3K9me3) | 0 | 0 |
| 0.1–1 | (Swi6HP1 + H3K9me3) | 1 | 0 | |
| 0.01–0.1 | (Swi6HP1 + H3K9me3) | 23 | 20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.B.; Belyakin, S.N.; Laktionov, P.P. Biology and Physics of Heterochromatin-Like Domains/Complexes. Cells 2020, 9, 1881. https://doi.org/10.3390/cells9081881
Singh PB, Belyakin SN, Laktionov PP. Biology and Physics of Heterochromatin-Like Domains/Complexes. Cells. 2020; 9(8):1881. https://doi.org/10.3390/cells9081881
Chicago/Turabian StyleSingh, Prim B., Stepan N. Belyakin, and Petr P. Laktionov. 2020. "Biology and Physics of Heterochromatin-Like Domains/Complexes" Cells 9, no. 8: 1881. https://doi.org/10.3390/cells9081881
APA StyleSingh, P. B., Belyakin, S. N., & Laktionov, P. P. (2020). Biology and Physics of Heterochromatin-Like Domains/Complexes. Cells, 9(8), 1881. https://doi.org/10.3390/cells9081881