Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Reagents and Antibodies
2.3. Plasmids
2.4. Preparation of HBV
2.5. HBV Infection of HepG2-hNTCP Cells and Their Transient Transfection
2.6. Transient Transfection
2.7. Sample Preparation
2.8. Co-Immunoprecipitation
2.9. Western Blot
2.10. HBeAg Detection by an Enzyme-Linked Immunosorbent Assay
2.11. Liquid Chromatography-Tandem Mass Spectrometry Analysis
2.12. Statistical Analysis
3. Results
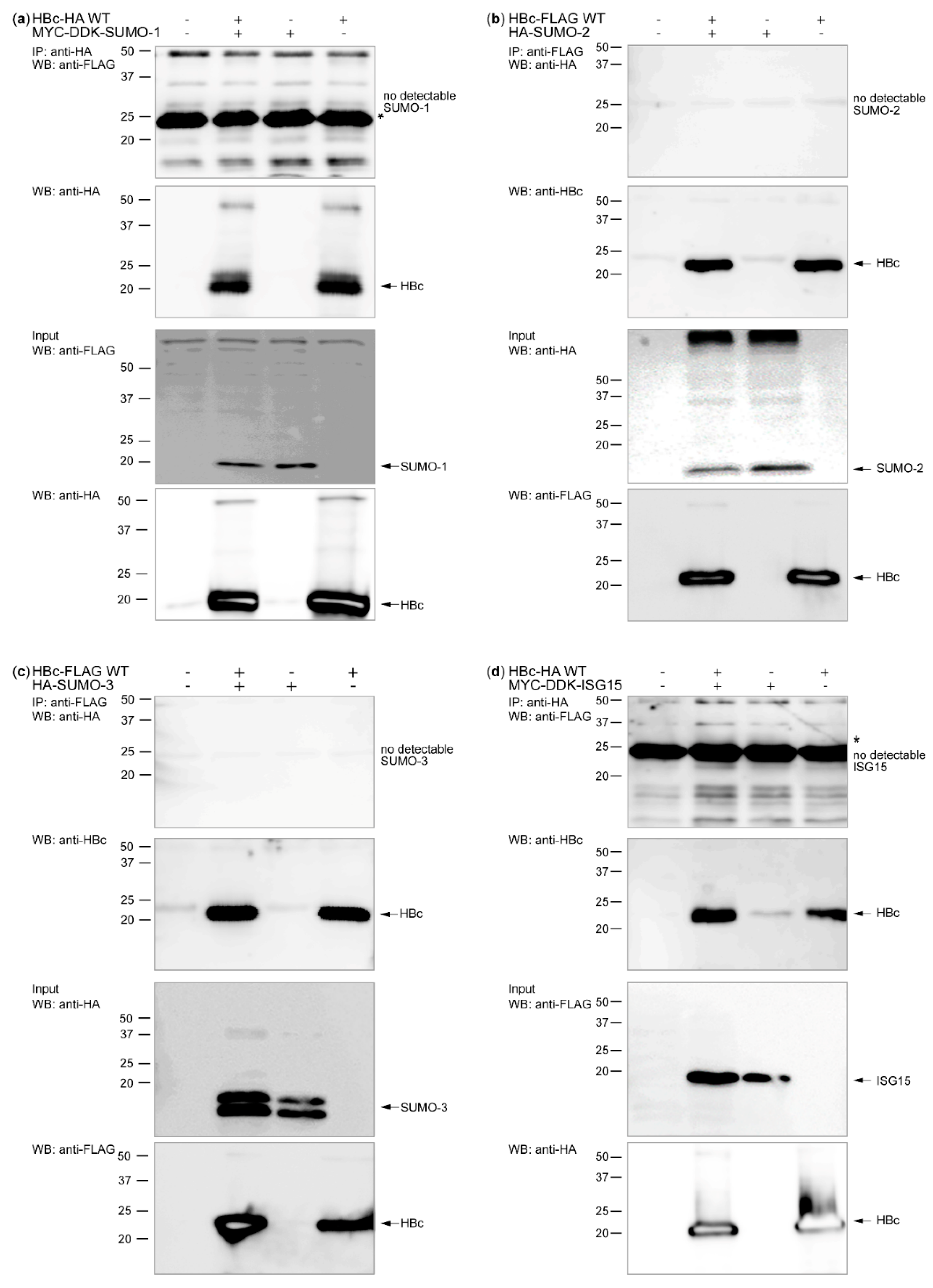
3.1. HBc and Its Post-Translational Modifiers
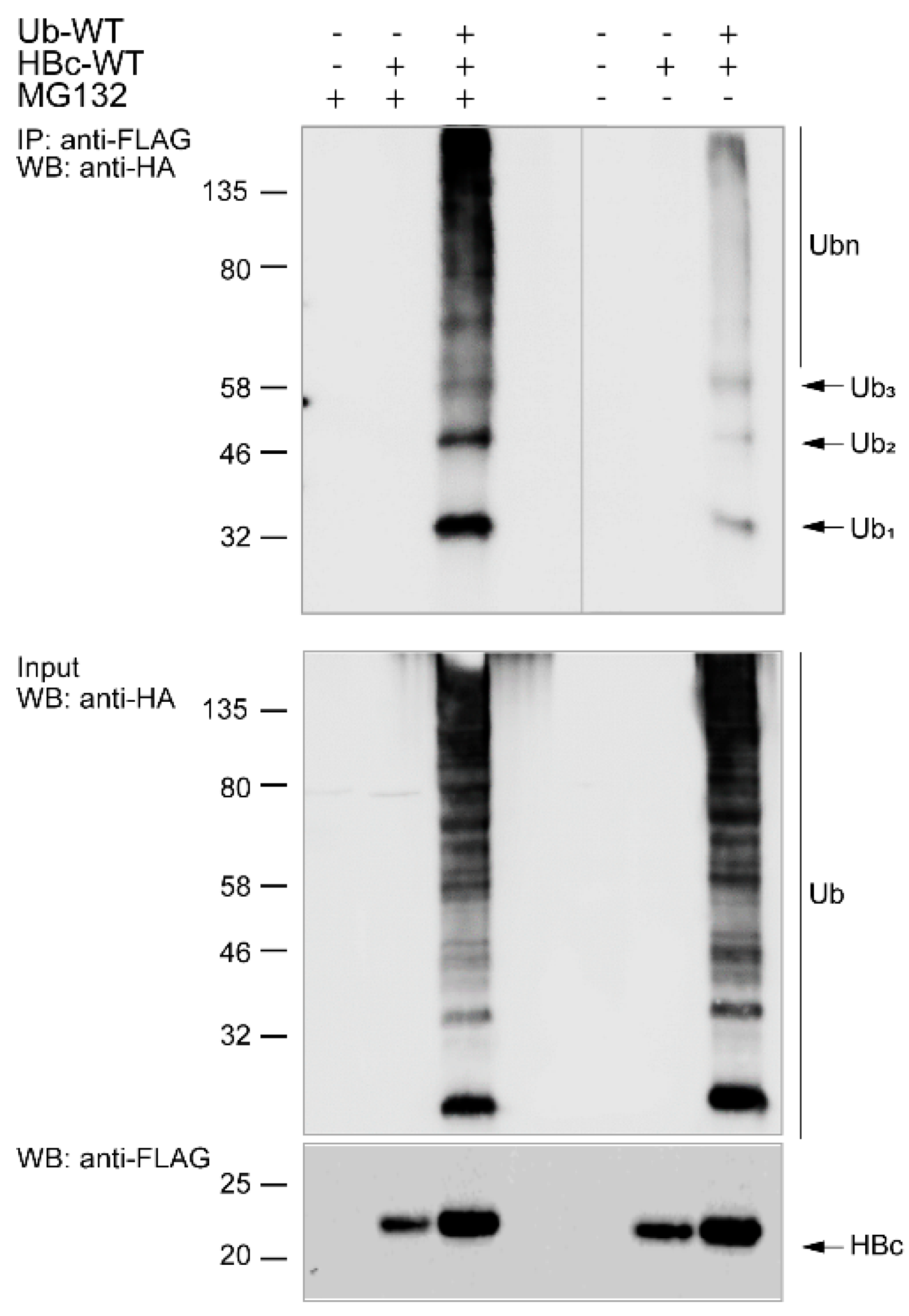
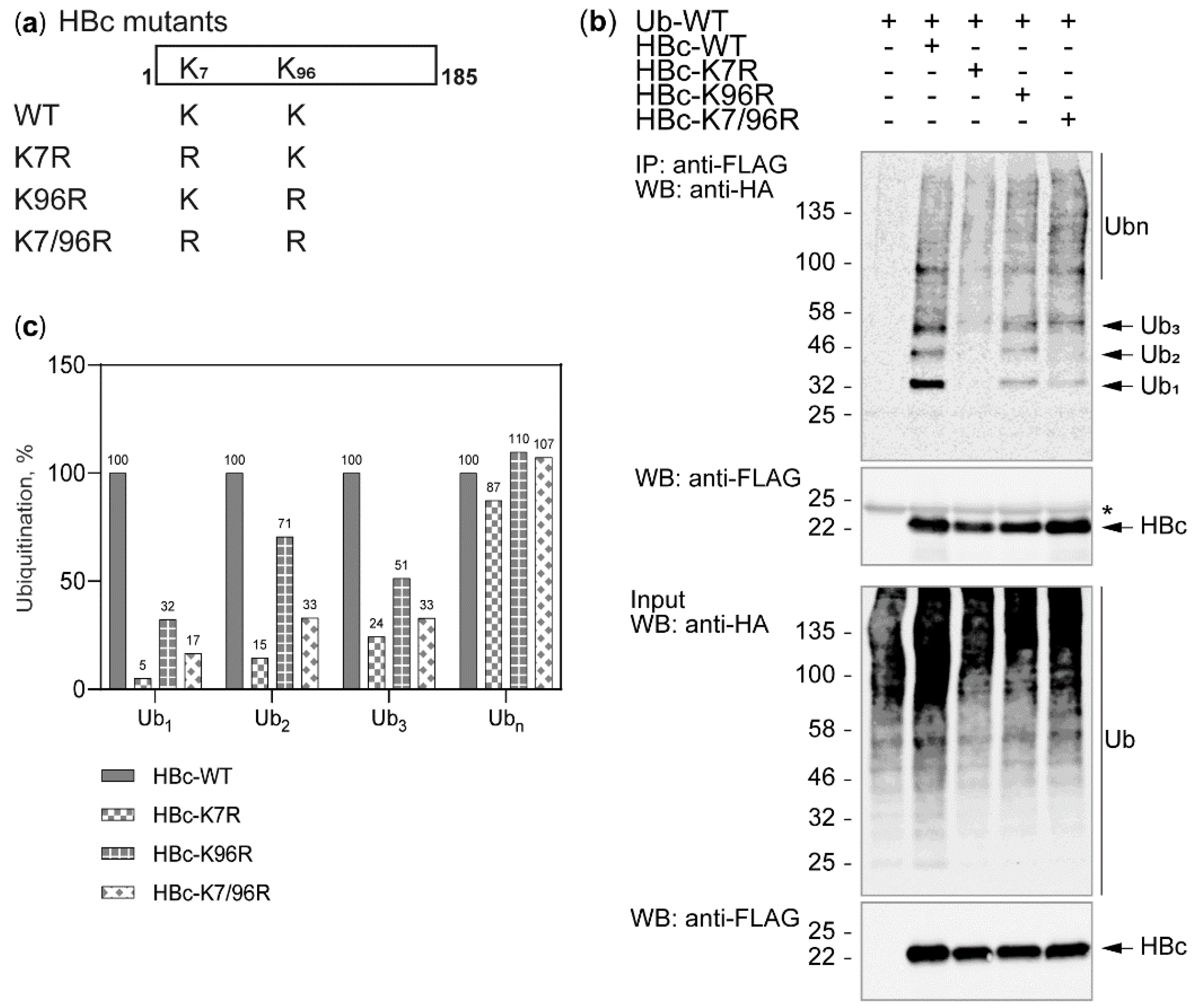
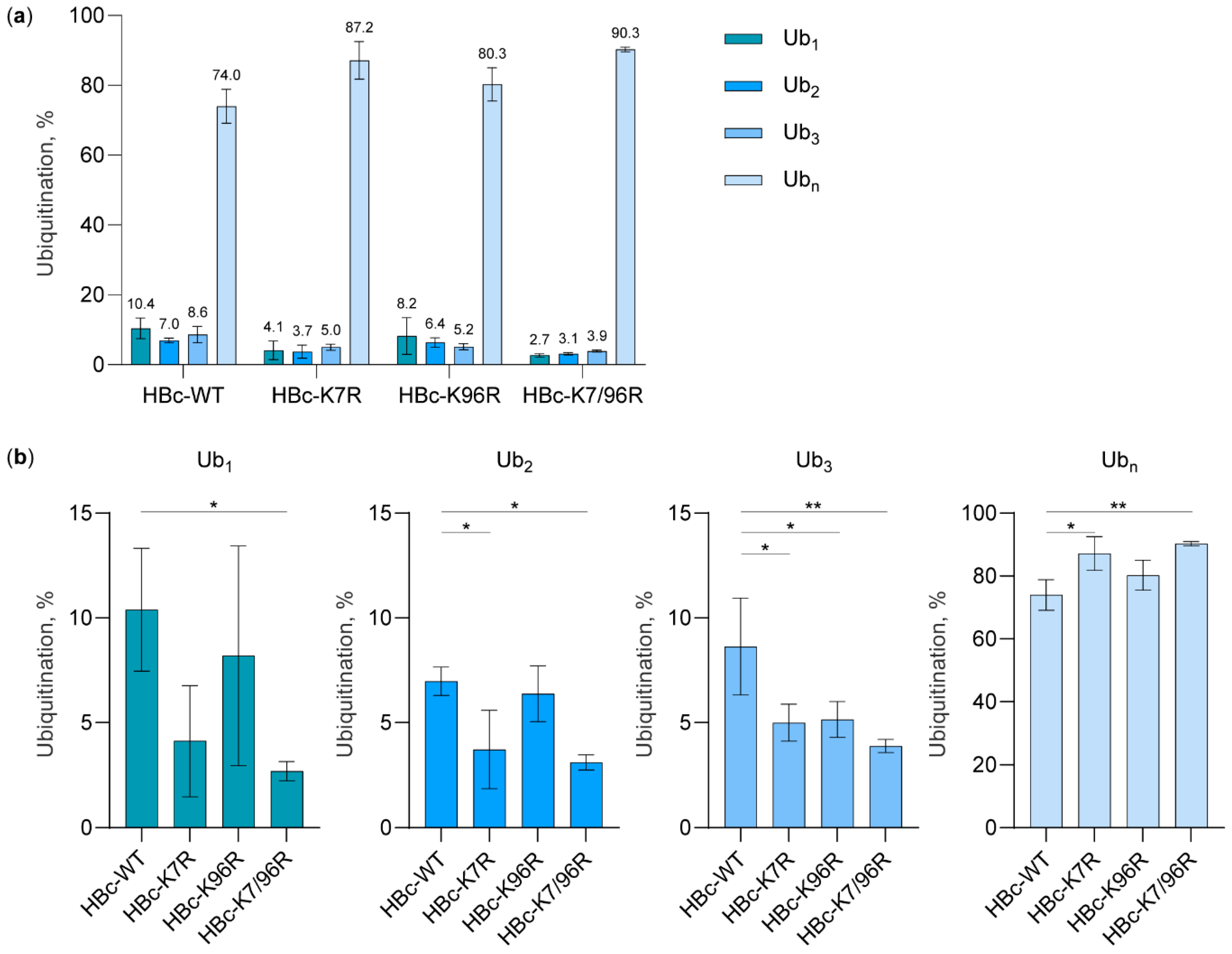
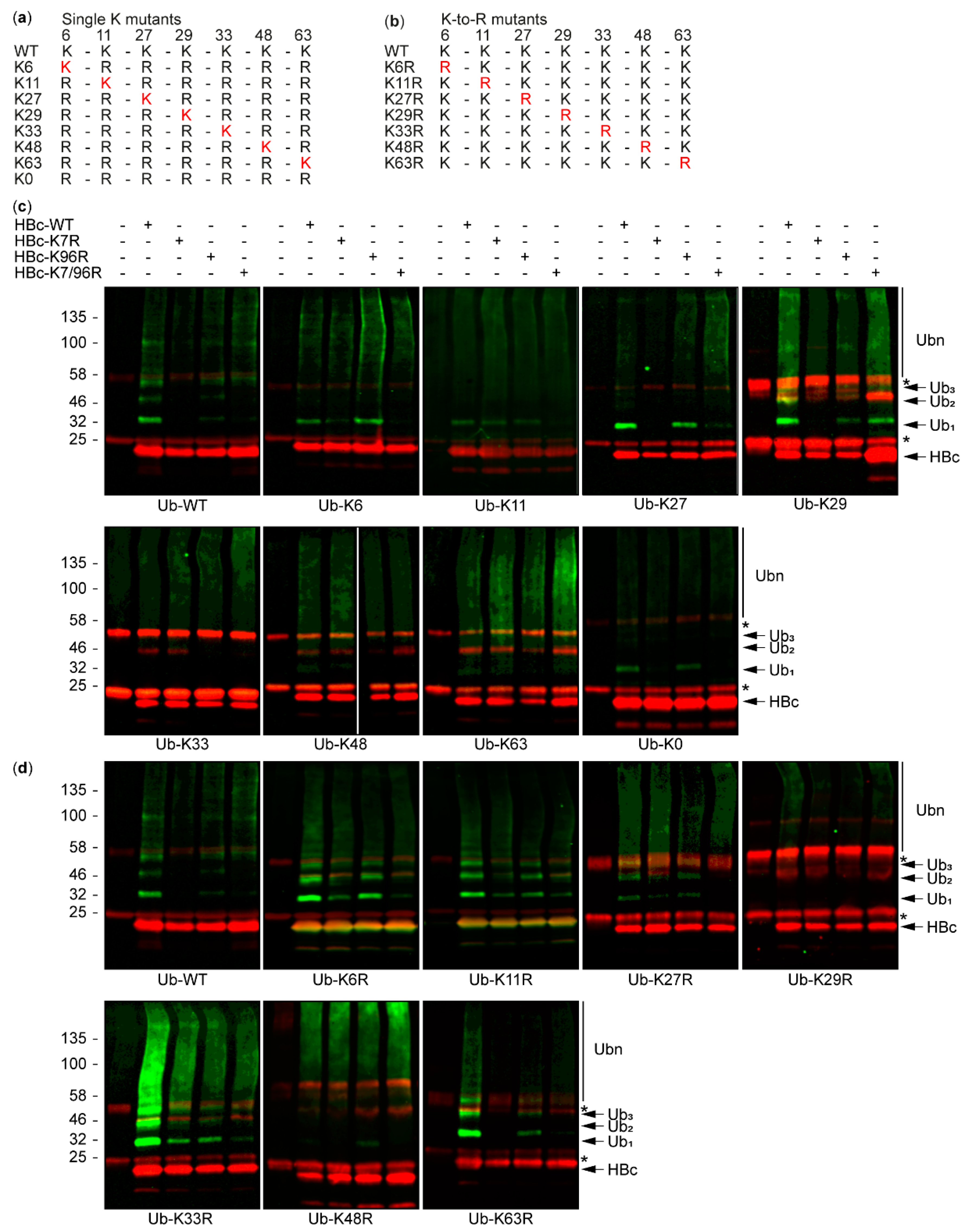
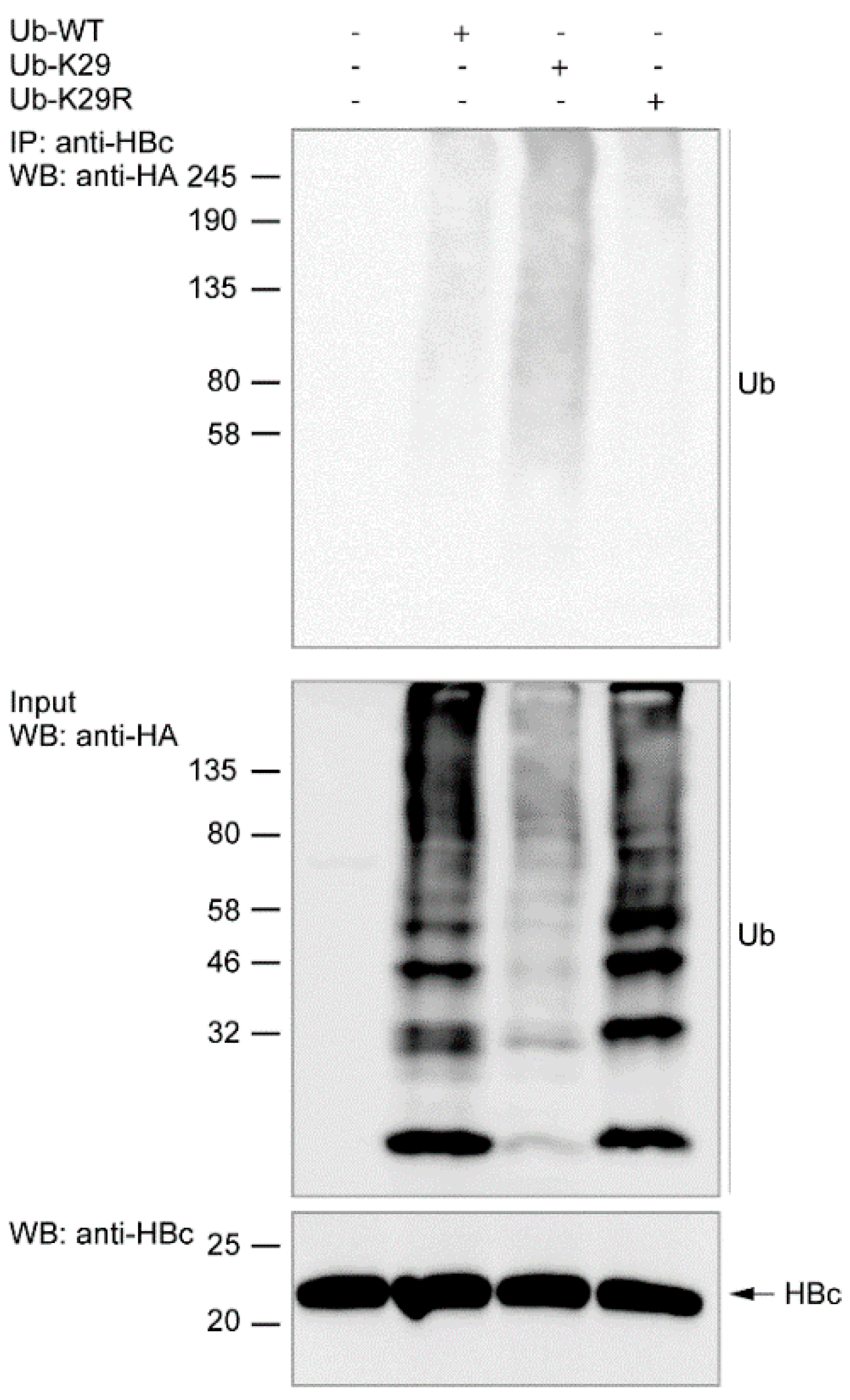
3.2. The Study of HBc Mutants and Ubiquitin Mutants
3.3. Ubiquitination of Endogenous HBc
3.4. MS Analysis of Ubiquitin-Protein Ligases Potentially Involved in HBc Ubiquitination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaefer, S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World. J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Schneider, R. Hepadnaviridae: The viruses and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 4, pp. 2923–2969. [Google Scholar]
- Hollinger, F.; Liang, T. Hepatitis B virus. In Fields Virology; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 4, pp. 2971–3036. [Google Scholar]
- Chain, B.M.; Myers, R. Variability and conservation in hepatitis B virus core protein. BMC Microbiol. 2005, 5, 33. [Google Scholar] [CrossRef]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef]
- Birnbaum, F.; Nassal, M. Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [Google Scholar] [CrossRef]
- Yang, F. Post-translational Modification Control of HBV Biological Processes. Front. Microbiol. 2018, 9, 2661. [Google Scholar] [CrossRef]
- Xuan, Q.; Zhang, Y.X.; Liu, D.G.; Chan, P.; Xu, S.L.; Cui, Y.Q. Post-translational modifications of α-synuclein contribute to neurodegeneration in the colon of elderly individuals. Mol. Med. Rep. 2016, 13, 5077–5083. [Google Scholar] [CrossRef]
- Chang, E.; Abe, J.I. Kinase-SUMO networks in diabetes-mediated cardiovascular disease. Metabolism 2016, 65, 623–633. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Ciechanover, A.; Merbl, Y. Post-translational modification profiling - A novel tool for mapping the protein modification landscape in cancer. Exp. Biol. Med. 2016, 241, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. Post-translational modifications in host cells during bacterial infection. FEBS Lett. 2010, 584, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar]
- Hershko, A.; Ciechanover, A. Mechanisms of intracellular protein breakdown. Annu. Rev. Biochem. 1982, 51, 335–364. [Google Scholar] [CrossRef]
- Ciechanover, A.; Elias, S.; Heller, H.; Hershko, A. “Covalent affinity” purification of ubiquitin-activating enzyme. J. Biol. Chem. 1982, 257, 2537–2542. [Google Scholar]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell. Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef]
- Wang, X.; Herr, R.A.; Chua, W.-J.; Lybarger, L.; Wiertz, E.J.H.J.; Hansen, T.H. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell. Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef]
- Ishikura, S.; Weissman, A.M.; Bonifacino, J.S. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J. Biol. Chem. 2010, 285, 23916–23924. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, J.; Jozwiak, A.A.; Hu, W.; Swiderski, P.M.; Chen, Y. Stability of thioester intermediates in ubiquitin-like modifications. Protein Sci. 2009, 18, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Pinto, M.P.; Grou, C.P.; Alencastre, I.S.; Fransen, M.; Sá-Miranda, C.; Azevedo, J.E. Ubiquitination of mammalian Pex5p, the peroxisomal import receptor. J. Biol. Chem. 2007, 282, 31267–31272. [Google Scholar] [CrossRef] [PubMed]
- Kragt, A.; Voorn-Brouwer, T.; van den Berg, M.; Distel, B. The Saccharomyces cerevisiae peroxisomal import receptor Pex5p is monoubiquitinated in wild type cells. J. Biol. Chem. 2005, 280, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.-J.; Rape, M. Enhanced Protein Degradation by Branched Ubiquitin Chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef]
- Rost, M.; Mann, S.; Lambert, C.; Döring, T.; Thomé, N.; Prange, R. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 2006, 281, 29297–29308. [Google Scholar] [CrossRef]
- Garcia, M.L.; Byfield, R.; Robek, M.D. Hepatitis B virus replication and release are independent of core lysine ubiquitination. J. Virol. 2009, 83, 4923–4933. [Google Scholar] [CrossRef][Green Version]
- Qian, G.; Jin, F.; Chang, L.; Yang, Y.; Peng, H.; Duan, C. NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation. Biotechnol. Lett. 2012, 34, 29–36. [Google Scholar] [CrossRef]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Lubyová, B.; Weber, J. Posttranslational modifications of HBV core protein. Acta. Virol. 2020, 64, 177–186. [Google Scholar] [CrossRef]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Chew, K.C.; Tan, J.M.; Wang, C.; Chung, K.K.; Zhang, Y.; Tanaka, Y.; Smith, W.; Engelender, S.; Ross, C.A.; et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: Implications for Lewy body formation. J. Neurosci. 2005, 25, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Livingston, C.M.; Ifrim, M.F.; Cowan, A.E.; Weller, S.K. Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.; Wu, H.C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef]
- Békés, M.; Prudden, J.; Srikumar, T.; Raught, B.; Boddy, M.N.; Salvesen, G.S. The dynamics and mechanism of SUMO chain deconjugation by SUMO-specific proteases. J. Biol. Chem. 2011, 286, 10238–10247. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Nguyen, H.P.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 3117–3120. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Choo, Y.S.; Zhang, Z. Detection of protein ubiquitination. J. Vis. Exp. 2009, 1293. [Google Scholar] [CrossRef]
- Chastagner, P.; Israël, A.; Brou, C. Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 2006, 7, 1147–1153. [Google Scholar] [CrossRef]
- Hay-Koren, A.; Caspi, M.; Zilberberg, A.; Rosin-Arbesfeld, R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates beta-catenin. Mol. Biol. Cell 2011, 22, 399–411. [Google Scholar] [CrossRef]
- Ponsel, D.; Bruss, V. Mapping of Amino Acid Side Chains on the Surface of Hepatitis B Virus Capsids Required for Envelopment and Virion Formation. J. Virol. 2003, 77, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.W.; Kovary, K.M.; Guillaume, J.; Chen, L.-c.; Teruel, M.N.; Wandless, T.J. The E3 ubiquitin ligase UBE3C enhances proteasome processivity by ubiquitinating partially proteolyzed substrates. J. Biol. Chem. 2013, 288, 34575–34587. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.N.; Ng, A.H.M.; Measday, V.; Mayor, T. Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 2011, 13, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl. Acad. Sci. USA 2018, 115, E1401–E1408. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell. Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Michel, M.A.; Elliott, P.R.; Swatek, K.N.; Simicek, M.; Pruneda, J.N.; Wagstaff, J.L.; Freund, S.M.V.; Komander, D. Assembly and Specific Recognition of K29- and K33-Linked Polyubiquitin. Mol. Cell 2015, 58, 95–109. [Google Scholar] [CrossRef]
- Bremm, A.; Freund, S.M.; Komander, D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol. 2010, 17, 939–947. [Google Scholar] [CrossRef]
- Kristariyanto, Y.A.; Choi, S.Y.; Rehman, S.A.; Ritorto, M.S.; Campbell, D.G.; Morrice, N.A.; Toth, R.; Kulathu, Y. Assembly and structure of Lys33-linked polyubiquitin reveals distinct conformations. Biochem. J. 2015, 467, 345–352. [Google Scholar] [CrossRef]
- Lee, T.L.; Shyu, Y.C.; Hsu, T.Y.; Shen, C.K. Itch regulates p45/NF-E2 in vivo by Lys63-linked ubiquitination. Biochem. Biophys. Res. Commun. 2008, 375, 326–330. [Google Scholar] [CrossRef]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, P.; Israël, A.; Brou, C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE 2008, 3, e2735. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Yeung, B.; Ho, K.-C.; Yang, X. WWP1 E3 Ligase Targets LATS1 for Ubiquitin-Mediated Degradation in Breast Cancer Cells. PLoS ONE 2013, 8, e61027. [Google Scholar] [CrossRef] [PubMed]
- Pao, K.C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; van Aalten, D.M.F.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef]
- Yin, L.; Joshi, S.; Wu, N.; Tong, X.; Lazar, M.A. E3 ligases Arf-bp1 and Pam mediate lithium-stimulated degradation of the circadian heme receptor Rev-erb alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 11614–11619. [Google Scholar] [CrossRef]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef]
- Karim, M.; Biquand, E.; Declercq, M.; Jacob, Y.; van der Werf, S.; Demeret, C. Nonproteolytic K29-Linked Ubiquitination of the PB2 Replication Protein of Influenza A Viruses by Proviral Cullin 4-Based E3 Ligases. mBio 2020, 11, e00305–e00320. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef]
- Narahara, C.; Yasuda, J. Roles of the three L-domains in β-retrovirus budding. Microbiol. Immunol. 2015, 59, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Fisher, R.D.; Chung, H.Y.; Myszka, D.G.; Sundquist, W.I.; Hill, C.P. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol. 2008, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Joshi, A.; Nagashima, K.; Freed, E.O.; Hurley, J.H. Structural basis for viral late-domain binding to Alix. Nat. Struct. Mol. Biol. 2007, 14, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Puffer, B.A.; Parent, L.J.; Wills, J.W.; Montelaro, R.C. Equine infectious anemia virus utilizes a YXXL motif within the late assembly domain of the Gag p9 protein. J. Virol. 1997, 71, 6541–6546. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.W.; Cameron, C.E.; Wilson, C.B.; Xiang, Y.; Bennett, R.P.; Leis, J. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 1994, 68, 6605–6618. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-F.; Tsai, M.-L.; Huang, J.-Y.; Chang, Y.-S.; Shih, C. The Dual Role of an ESCRT-0 Component HGS in HBV Transcription and Naked Capsid Secretion. PLoS Pathog. 2015, 11, e1005123. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Addgene Plasmid | Citation | Reference |
|---|---|---|
| pRK5-HA-Ubiquitin-WT | a gift from Ted Dawson (Addgene plasmid #17608; http://n2t.net/addgene:17608; RRID:Addgene_17608) | [34] |
| pRK5-HA-Ubiquitin-K6 | a gift from Sandra Weller (Addgene plasmid #22900; http://n2t.net/addgene:22900; RRID:Addgene_22900) | [35] |
| pRK5-HA-Ubiquitin-K11 | a gift from Sandra Weller (Addgene plasmid #22901; http://n2t.net/addgene:22901; RRID:Addgene_22901) | [35] |
| pRK5-HA-Ubiquitin-K27 | a gift from Sandra Weller (Addgene plasmid #22902; http://n2t.net/addgene:22902; RRID:Addgene_22902) | [35] |
| pRK5-HA-Ubiquitin-K29 | a gift from Sandra Weller (Addgene plasmid #22903; http://n2t.net/addgene:22903; RRID:Addgene_22903) | [35] |
| pRK5-HA-Ubiquitin-K33 | a gift from Ted Dawson (Addgene plasmid #17607; http://n2t.net/addgene:17607; RRID:Addgene_17607) | [34] |
| pRK5-HA-Ubiquitin-K48 | a gift from Ted Dawson (Addgene plasmid #17605; http://n2t.net/addgene:17605; RRID:Addgene_17605) | [34] |
| pRK5-HA-Ubiquitin-K63 | a gift from Ted Dawson (Addgene plasmid #17606; http://n2t.net/addgene:17606; RRID:Addgene_17606) | [34] |
| pRK5-HA-Ubiquitin-K0 | a gift from Ted Dawson (Addgene plasmid #17603; http://n2t.net/addgene:17603; RRID:Addgene_17603) | [34] |
| pRK5-HA-Ubiquitin-K6R | a gift from Josef Kittler (Addgene plasmid #121153; http://n2t.net/addgene:121153; RRID:Addgene_121153) | [36] |
| pRK5-HA-Ubiquitin-K11R | a gift from Josef Kittler (Addgene plasmid #121154; http://n2t.net/addgene:121154; RRID:Addgene_121154) | [36] |
| pRK5-HA-Ubiquitin-K27R | a gift from Josef Kittler (Addgene plasmid #121155; http://n2t.net/addgene:121155; RRID:Addgene_121155) | [36] |
| pRK5-HA-Ubiquitin-K29R | a gift from Ted Dawson (Addgene plasmid #17602; http://n2t.net/addgene:17602; RRID:Addgene_17602) | [34] |
| pRK5-HA-Ubiquitin-K48R | a gift from Ted Dawson (Addgene plasmid #17604; http://n2t.net/addgene:17604; RRID:Addgene_17604) | [34] |
| pcDNA3 HA-SUMO2 WT | a gift from Guy Salvesen (Addgene plasmid #48967; http://n2t.net/addgene:48967; RRID:Addgene_48967) | [37] |
| pcDNA3/HA-SUMO3 (Sentrin 2) | a gift from Edward Yeh (Addgene plasmid #17361; http://n2t.net/addgene:17361; RRID:Addgene_17361) | [38] |
| E3 Ubiquitin-Protein Ligase | HBc # Peptides Exp1/Exp2/Exp3 | HBc # PSMs Exp1/Exp2/Exp3 | CTRL # Peptides Exp1/Exp2/Exp3 | CTRL # PSMs Exp1/Exp2/Exp3 |
|---|---|---|---|---|
| Baculoviral IAP repeat-containing protein 6 BIRC6 * | 122/109/124 | 219/171/204 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase UBR4 * | 96/75/90 | 140/101/121 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase HUWE1 * | 78/67/84 | 125/90/117 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase UBR5 * | 43/30/38 | 59/40/52 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase TRIM21 | 26/24/26 | 57/51/47 | 26/24/21 | 62/49/50 |
| E3 ubiquitin-protein ligase HECTD1 * | 24/7/14 | 25/7/15 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase UBR2 * | 17/6/12 | 19/8/12 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase TRIM71 | 7/2/4 | 9/2/4 | 1/0/0 | 1/0/0 |
| Ubiquitin-protein ligase E3A | 6/3/4 | 8/3/4 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase MARCH7 | 5/2/0 | 5/2/0 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase TRIP12 | 5/3/4 | 6/3/5 | 0/0/0 | 0/0/0 |
| NEDD4-like E3 ubiquitin-protein ligase WWP1 | 5/1/4 | 5/1/4 | 9/3/7 | 10/3/7 |
| E3 ubiquitin-protein ligase KCMF1 * | 4/5/7 | 6/6/9 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase RNF31 * | 4/4/2 | 4/4/2 | 1/0/0 | 1/0/0 |
| E3 ubiquitin-protein ligase Mdm2 * | 3/3/4 | 3/3/4 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase UBR1 * | 3/2/2 | 4/2/2 | 0/0/0 | 0/0/0 |
| Putative E3 ubiquitin-protein ligase SH3RF2 | 2/2/2 | 4/4/3 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase RNF138 | 2/1/2 | 2/1/2 | 2/1/0 | 2/1/0 |
| E3 ubiquitin-protein ligase RNF181 * | 2/1/0 | 2/1/0 | 0/0/0 | 0/0/0 |
| Probable E3 ubiquitin-protein ligase HERC1 | 2/0/0 | 3/0/0 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase MIB1 | 2/0/1 | 2/0/1 | 3/0/3 | 4/0/4 |
| NEDD4-like E3 ubiquitin-protein ligase WWP2 | 1/1/2 | 1/1/2 | 1/1/1 | 1/1/1 |
| E3 ubiquitin-protein ligase AMFR | 1/1/1 | 1/1/1 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase RING1 | 1/2/2 | 1/2/2 | 1/2/0 | 1/2/0 |
| E3 ubiquitin-protein ligase RBBP6 | 1/2/3 | 1/2/3 | 3/1/2 | 5/1/3 |
| E3 ubiquitin-protein ligase Praja-1 | 1/0/1 | 1/0/1 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase rififylin RFFL | 1/0/0 | 1/0/0 | 0/0/0 | 0/0/0 |
| E3 ubiquitin-protein ligase MYCBP2 | 0/0/3 | 0/0/3 | 0/0/0 | 0/0/0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langerová, H.; Lubyová, B.; Zábranský, A.; Hubálek, M.; Glendová, K.; Aillot, L.; Hodek, J.; Strunin, D.; Janovec, V.; Hirsch, I.; et al. Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination. Cells 2020, 9, 2547. https://doi.org/10.3390/cells9122547
Langerová H, Lubyová B, Zábranský A, Hubálek M, Glendová K, Aillot L, Hodek J, Strunin D, Janovec V, Hirsch I, et al. Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination. Cells. 2020; 9(12):2547. https://doi.org/10.3390/cells9122547
Chicago/Turabian StyleLangerová, Hana, Barbora Lubyová, Aleš Zábranský, Martin Hubálek, Kristýna Glendová, Ludovic Aillot, Jan Hodek, Dmytro Strunin, Václav Janovec, Ivan Hirsch, and et al. 2020. "Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination" Cells 9, no. 12: 2547. https://doi.org/10.3390/cells9122547
APA StyleLangerová, H., Lubyová, B., Zábranský, A., Hubálek, M., Glendová, K., Aillot, L., Hodek, J., Strunin, D., Janovec, V., Hirsch, I., & Weber, J. (2020). Hepatitis B Core Protein Is Post-Translationally Modified through K29-Linked Ubiquitination. Cells, 9(12), 2547. https://doi.org/10.3390/cells9122547