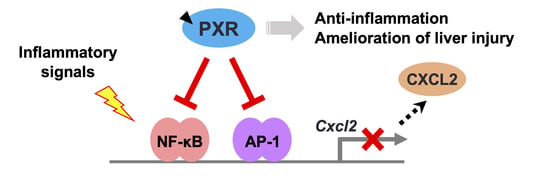
PXR Functionally Interacts with NF-κB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmid Preparation
2.3. Animal Experiments
2.4. Cell Culture
2.5. Liver Histology Analysis
2.6. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.7. Reporter Assays
2.8. Statistical Analysis
3. Results
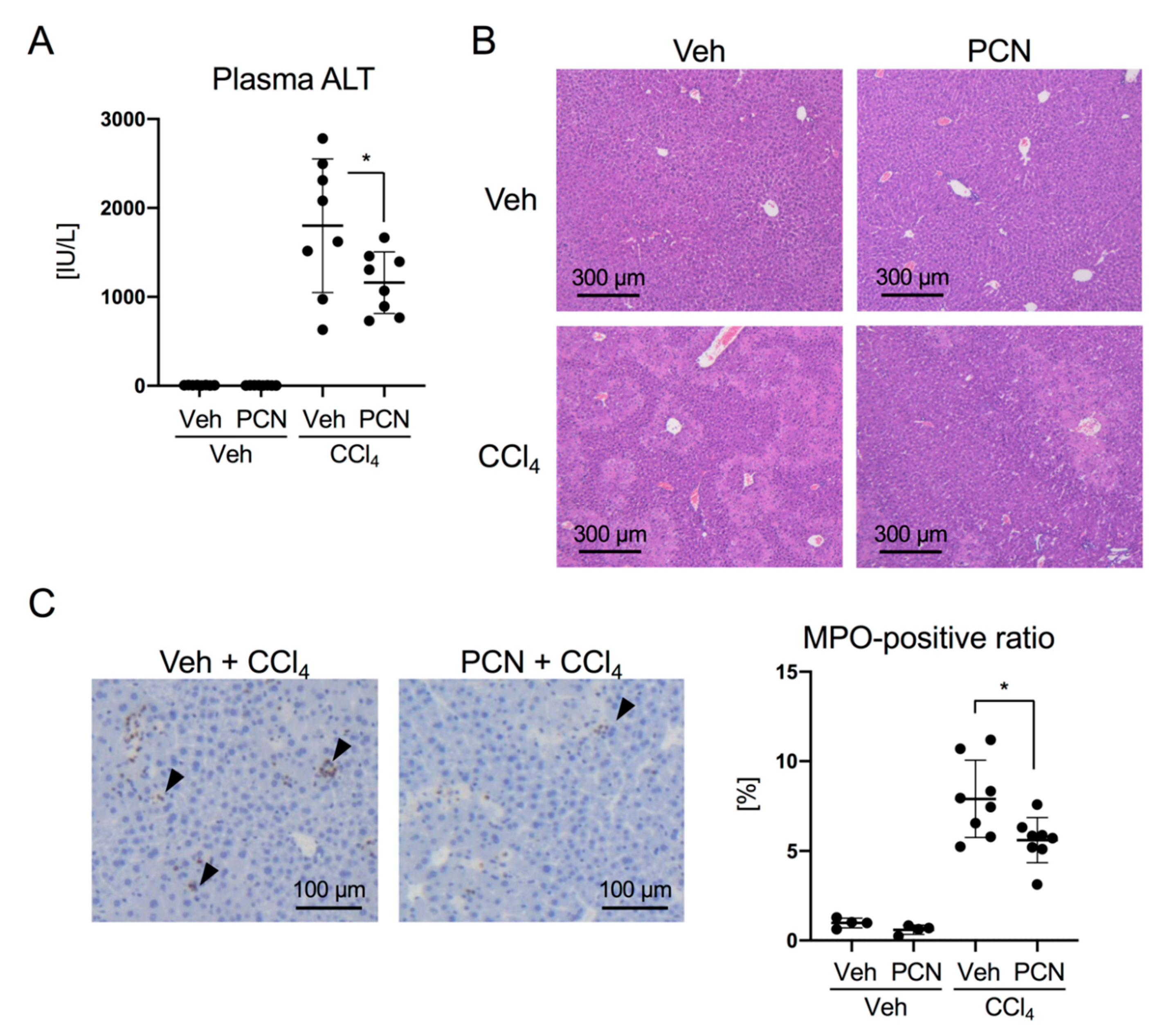
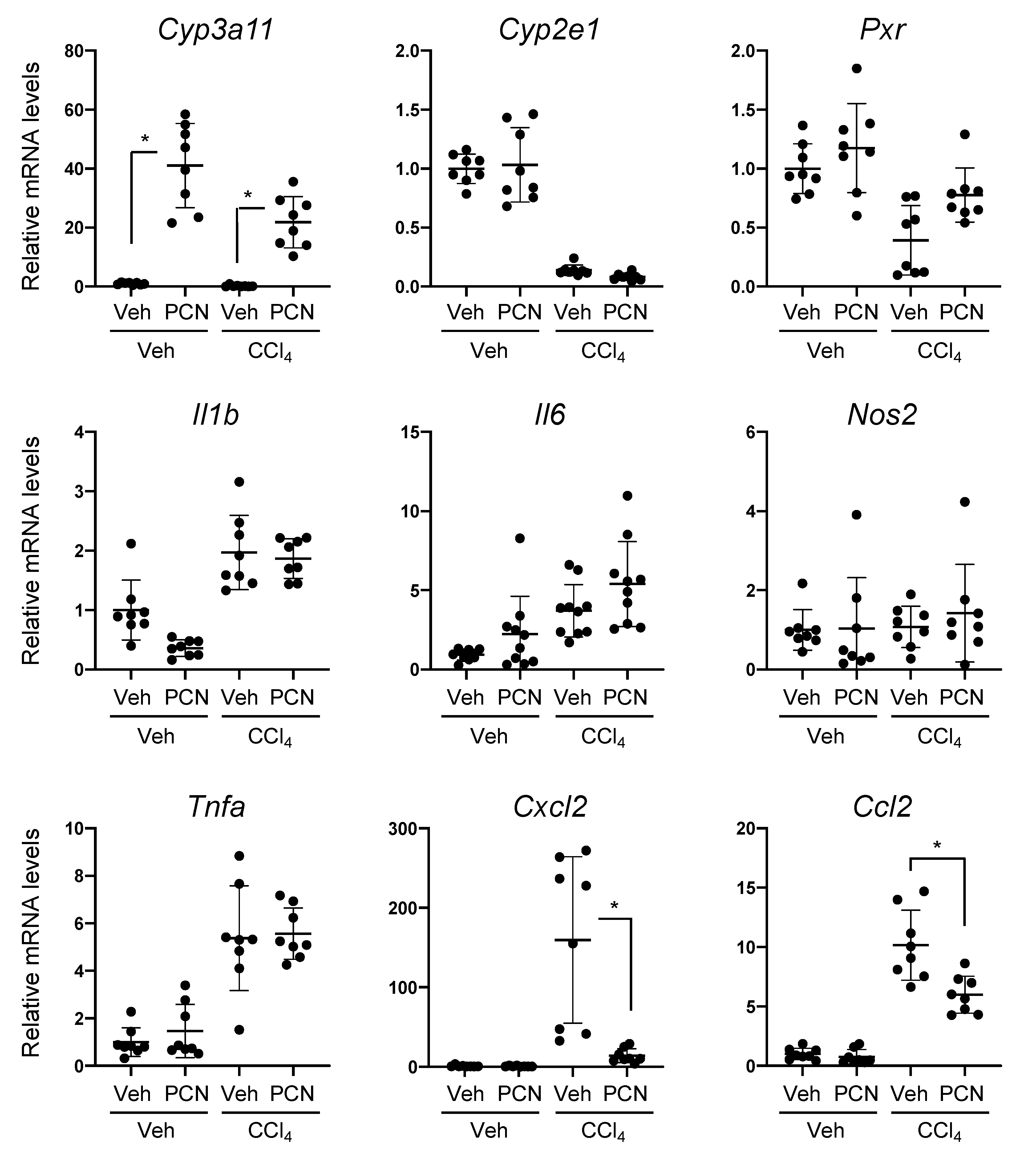
3.1. PCN Treatment Ameliorates Liver Injury-Induced Expression of Chemokines in Mice
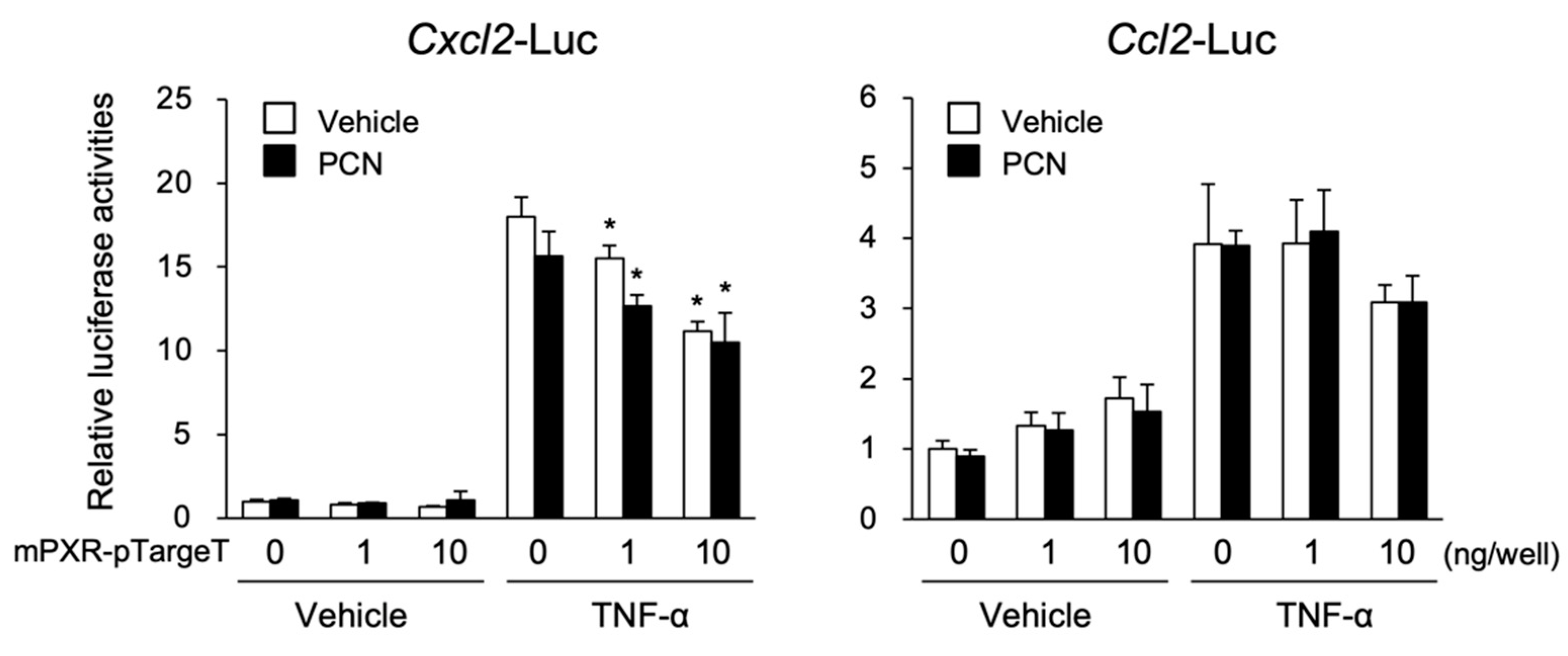
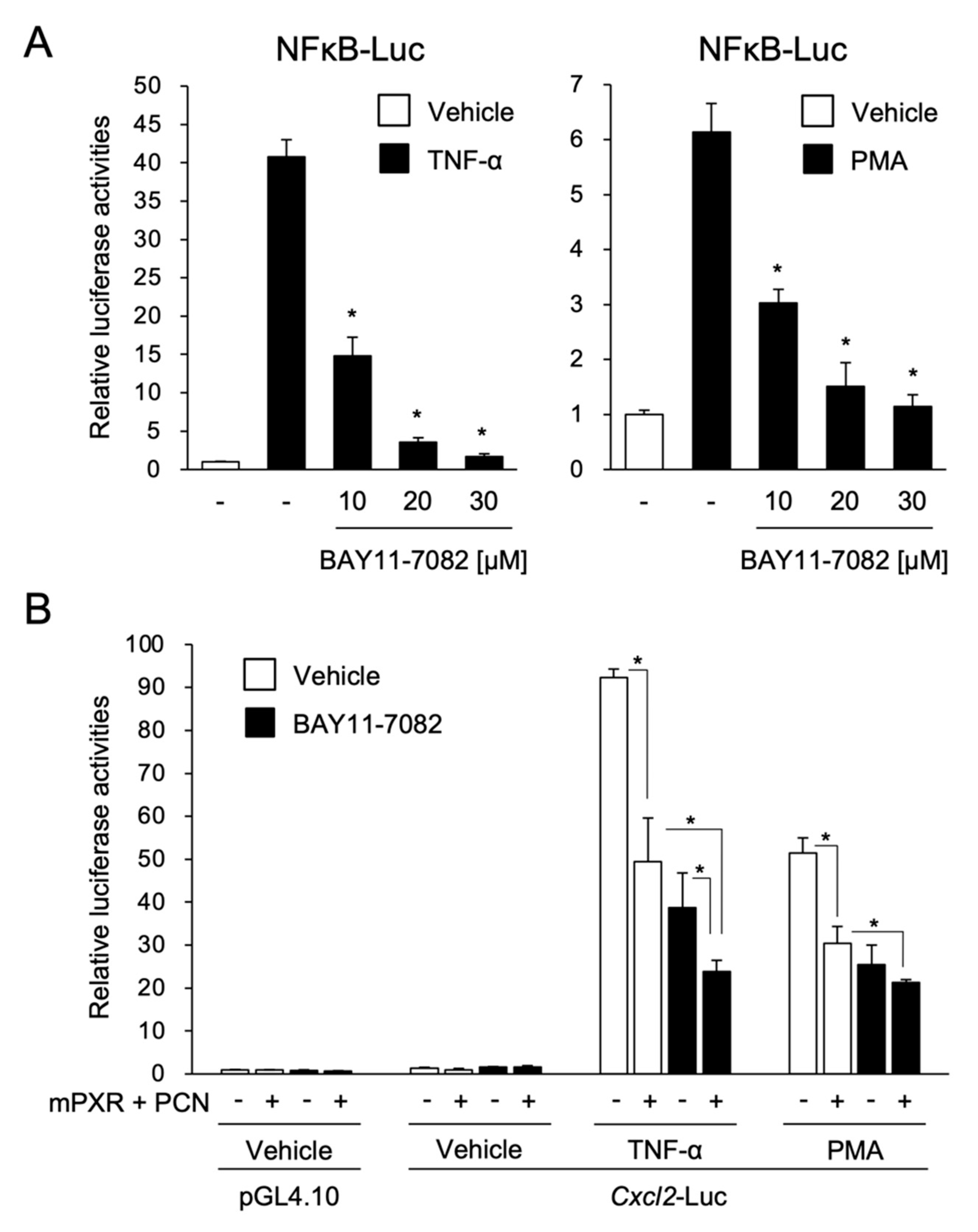
3.2. PXR Suppresses the NF-κB-Dependent Cxcl2 Expression
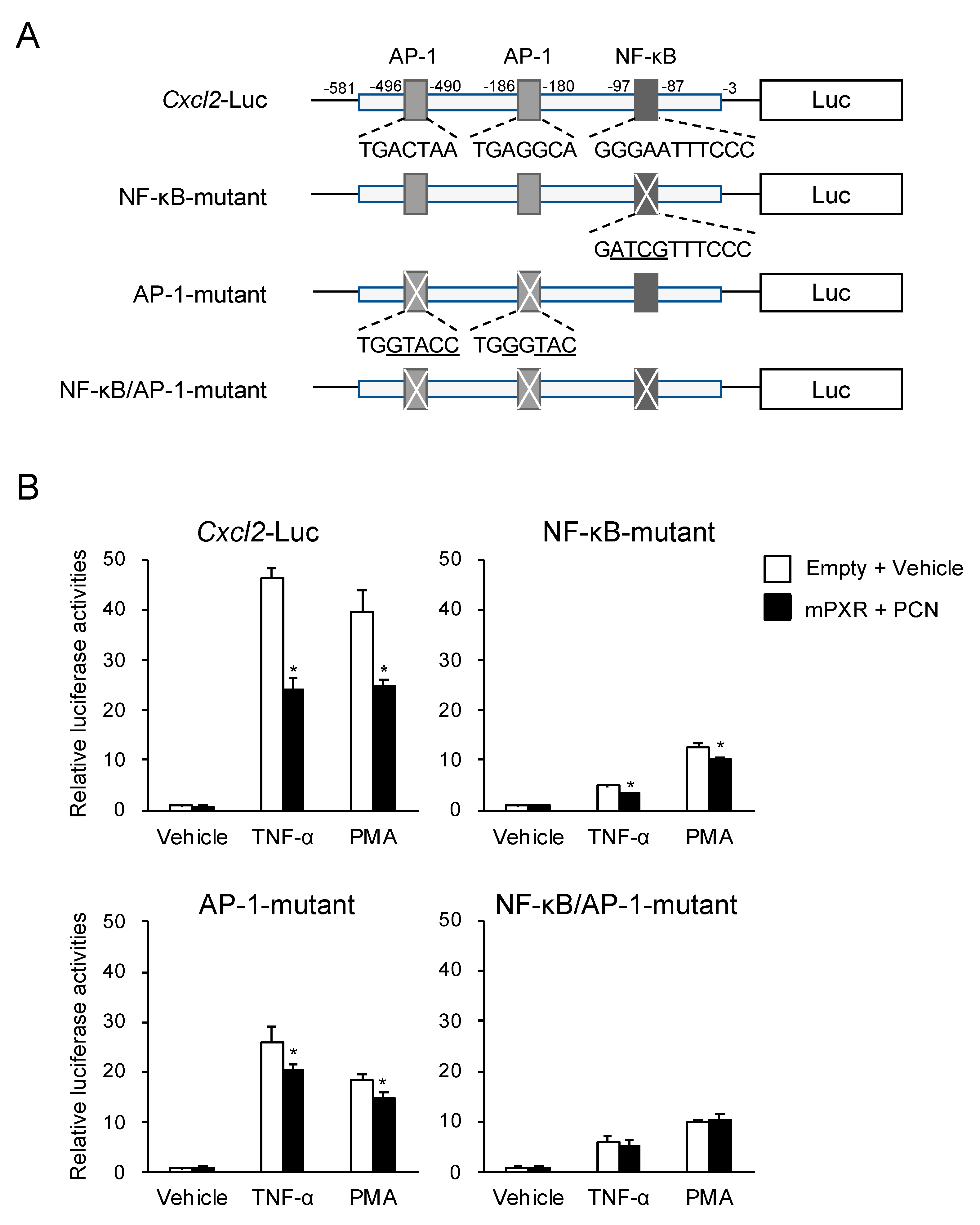
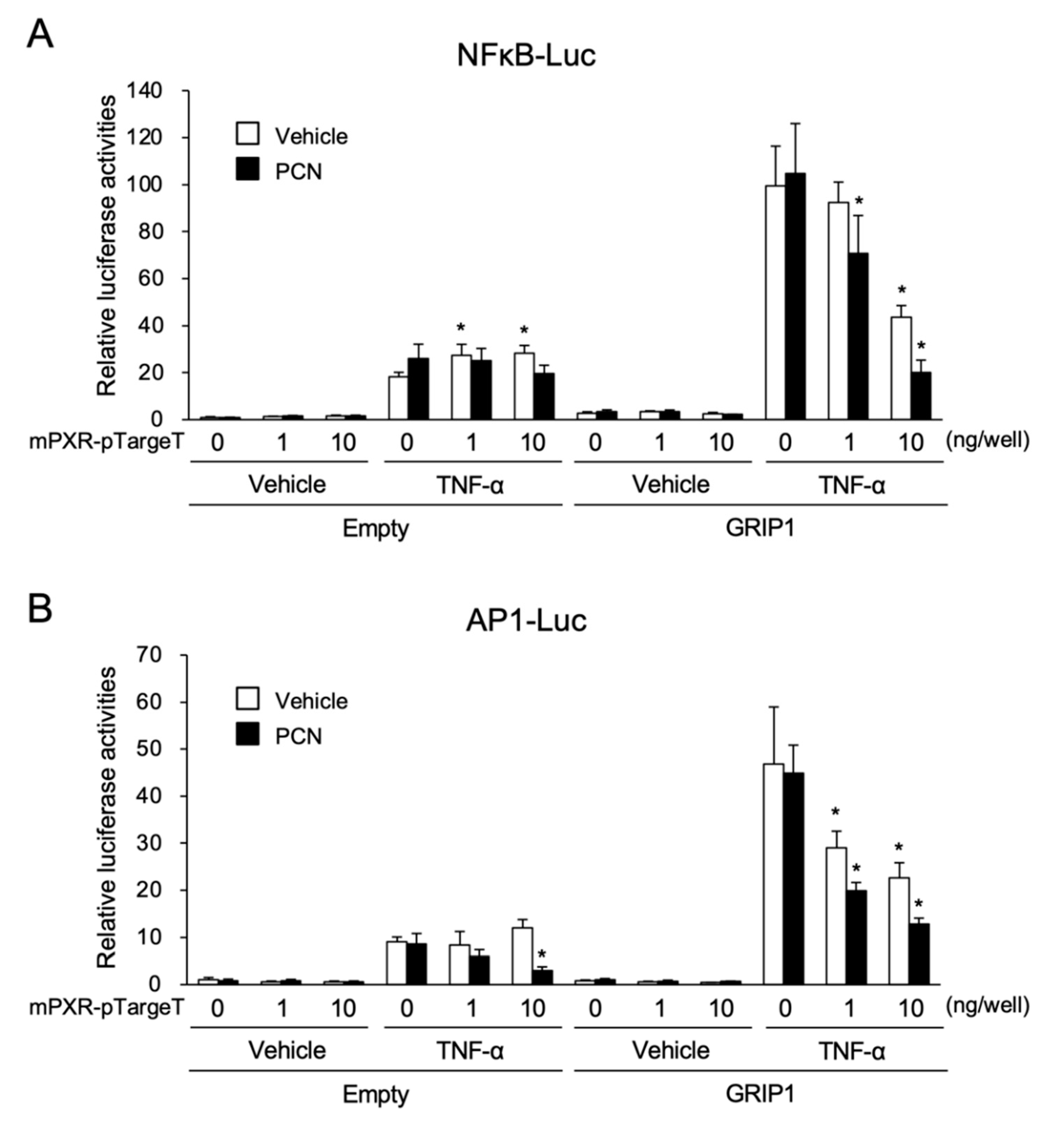
3.3. PXR Downregulates AP-1- and NF-κB-Dependent Cxcl2 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Koike, C.; Negishi, M.; Yamamoto, Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol. Cell. Biol. 2004, 24, 7931–7940. [Google Scholar] [CrossRef]
- Gotoh, S.; Negishi, M. Serum- and glucocorticoid-regulated kinase 2 determines drug-activated pregnane X receptor to induce gluconeogenesis in human liver cells. J. Pharmacol. Exp. Ther. 2014, 348, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Moore, R.; Yamamoto, Y.; Negishi, M. Human nuclear pregnane X receptor cross-talk with CREB to repress cAMP activation of the glucose-6-phosphatase gene. Biochem. J. 2007, 407, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Shizu, R.; Abe, T.; Benoki, S.; Takahashi, M.; Kodama, S.; Miayata, M.; Matsuzawa, A.; Yoshinari, K. PXR stimulates growth factor-mediated hepatocyte proliferation by cross-talk with the FOXO transcription factor. Biochem. J. 2016, 473, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Shizu, R.; Sasaki, T.; Shimizu, Y.; Hosaka, T.; Kodama, S.; Matsuzawa, A.; Yoshinari, K. Functional interaction between pregnane X receptor and Yes-associated protein in xenobiotic-dependent liver hypertrophy and drug metabolism. J. Pharmacol. Exp. Ther. 2019, 371, 590–601. [Google Scholar] [CrossRef]
- Jiang, Y.; Feng, D.; Ma, X.; Fan, S.; Gao, Y.; Fu, K.; Wang, Y.; Sun, J.; Yao, X.; Liu, C.; et al. Pregnane X receptor regulates liver size and liver cell fate by Yes-associated protein activation in mice. Hepatology 2019, 69, 343–358. [Google Scholar] [CrossRef]
- Shizu, R.; Yoshinari, K. Nuclear receptor CAR-mediated liver cancer and its species differences. Expert. Opin. Drug Metab. Toxicol. 2020, 16, 343–351. [Google Scholar] [CrossRef]
- Yoshinari, K. Role of nuclear receptors PXR and CAR in xenobiotic-induced hepatocyte proliferation and chemical carcinogenesis. Biol. Pharm. Bull. 2019, 42, 1243–1252. [Google Scholar] [CrossRef]
- Hu, G.; Xu, C.; Staudinger, J.L. Pregnane X receptor is SUMOylated to repress the inflammatory response. J. Pharmacol. Exp. Ther. 2010, 335, 342–350. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Palladino, G.; Claudio, D.; Ricci, P.; Distrutti, E.; Barbanti, M.; Baldelli, F.; Fiorucci, S. Inhibition of NF-kappaB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur. J. Pharmacol. 2011, 668, 317–324. [Google Scholar] [CrossRef]
- Sun, M.; Cui, W.; Woody, S.K.; Staudinger, J.L. Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes. Drug Metab. Dispos. 2015, 43, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grün, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef]
- Kodama, S.; Shimura, T.; Kuribayashi, H.; Abe, T.; Yoshinari, K. Pregnenolone 16α-carbonitrile ameliorates concanavalin A-induced liver injury in mice independent of the nuclear receptor PXR activation. Toxicol. Lett. 2017, 271, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological relevance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Shizu, R.; Hosaka, T.; Sasaki, T.; Yoshinari, K. Possible involvement of the competition for the transcriptional coactivator glucocorticoid receptor-interacting protein 1 in the inflammatory signal-dependent suppression of PXR-mediated CYP3A induction in vitro. Drug Metab. Pharmacokinet. 2019, 34, 272–279. [Google Scholar] [CrossRef]
- Helsley, R.N.; Sui, Y.; Ai, N.; Park, S.H.; Welsh, W.J.; Zhou, C. Pregnane X receptor mediates dyslipidemia induced by the HIV protease inhibitor amprenavir in mice. Mol. Pharmacol. 2013, 83, 1190–1199. [Google Scholar] [CrossRef]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar] [CrossRef]
- Xie, W.; Tian, Y. Xenobiotic receptor meets NF-kappaB, a collision in the small bowel. Cell Metab. 2006, 4, 177–178. [Google Scholar] [CrossRef]
- Zhou, C.; Verma, S.; Blumberg, B. The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nuclear Recept. Signal 2009, 7, e001. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Ho Han, J.; Kwon, H.-J. NF-κB and c-Jun-dependent regulation of macrophage inflammatory protein-2 gene expression in response to lipopolysaccharide in RAW 264.7 cells. Mol. Immunol. 2003, 40, 633–643. [Google Scholar] [CrossRef]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Sorrell, T.C.; Forbes, I.J. Depression of immune competence by phenytoin and carbamazepine. Studies in vivo and in vitro. Clin. Exp. Immunol. 1975, 20, 273–285. [Google Scholar]
- Dou, W.; Zhang, J.; Zhang, E.; Sun, A.; Ding, L.; Chou, G.; Wang, Z.; Mani, S. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-κB signaling pathway. J. Pharmacol. Exp. Ther. 2013, 345, 473–482. [Google Scholar] [CrossRef]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Mencarelli, A.; D’Amore, C.; Renga, B.; Zampella, A.; Fiorucci, S. Total synthesis and pharmacological characterization of solomonsterol A, a potent marine pregnane-X-receptor agonist endowed with anti-inflammatory activity. J. Med. Chem. 2011, 54, 4590–4599. [Google Scholar] [CrossRef]
- Beck, I.M.E.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in inflammation: The interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef]
- McKay, L.I.; Cidlowski, J.A. Molecular control of immune/inflammatory responses: Interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr. Rev. 1999, 20, 435–459. [Google Scholar]
- Na, S.Y.; Kang, B.Y.; Chung, S.W.; Han, S.J.; Ma, X.; Trinchieri, G.; Im, S.Y.; Lee, J.W.; Kim, T.S. Retinoids inhibit interleukin-12 production in macrophages through physical associations of retinoid X receptor and NFkappaB. J. Biol. Chem. 1999, 274, 7674–7680. [Google Scholar] [CrossRef]
- Oladimeji, P.; Cui, H.; Zhang, C.; Chen, T. Regulation of PXR and CAR by protein-protein interaction and signaling crosstalk. Expert. Opin. Drug Metab. Toxicol. 2016, 12, 997–1010. [Google Scholar] [CrossRef]
- Lee, S.K.; Kim, H.J.; Na, S.Y.; Kim, T.S.; Choi, H.S.; Im, S.Y.; Lee, J.W. Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J. Biol. Chem. 1998, 273, 16651–16654. [Google Scholar] [CrossRef] [PubMed]
- Na, S.Y.; Lee, S.K.; Han, S.J.; Choi, H.S.; Im, S.Y.; Lee, J.W. Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor kappaB-mediated transactivations. J. Biol. Chem. 1998, 273, 10831–10834. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Na, S.Y.; Jung, S.Y.; Choi, J.E.; Jhun, B.H.; Cheong, J.; Meltzer, P.S.; Lee, Y.C.; Lee, J.W. Activating protein-1, nuclear factor-kappaB, and serum response factor as novel target molecules of the cancer-amplified transcription coactivator ASC-2. Mol. Endocrinol. 2000, 14, 915–925. [Google Scholar] [PubMed]
- Surapureddi, S.; Rana, R.; Goldstein, J.A. NCOA6 differentially regulates the expression of the CYP2C9 and CYP3A4 genes. Pharmacol. Res. 2011, 63, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, Y.; Liu, J.; Nie, Y.; Zhong, X.B.; Kan, Q.; Zhang, L. Alterations of histone modifications contribute to pregnane X receptor-mediated induction of CYP3A4 by rifampicin. Mol. Pharmacol. 2017, 92, 113–123. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamura, M.; Shizu, R.; Abe, T.; Kodama, S.; Hosaka, T.; Sasaki, T.; Yoshinari, K. PXR Functionally Interacts with NF-κB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells 2020, 9, 2296. https://doi.org/10.3390/cells9102296
Okamura M, Shizu R, Abe T, Kodama S, Hosaka T, Sasaki T, Yoshinari K. PXR Functionally Interacts with NF-κB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells. 2020; 9(10):2296. https://doi.org/10.3390/cells9102296
Chicago/Turabian StyleOkamura, Maya, Ryota Shizu, Taiki Abe, Susumu Kodama, Takuomi Hosaka, Takamitsu Sasaki, and Kouichi Yoshinari. 2020. "PXR Functionally Interacts with NF-κB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice" Cells 9, no. 10: 2296. https://doi.org/10.3390/cells9102296
APA StyleOkamura, M., Shizu, R., Abe, T., Kodama, S., Hosaka, T., Sasaki, T., & Yoshinari, K. (2020). PXR Functionally Interacts with NF-κB and AP-1 to Downregulate the Inflammation-Induced Expression of Chemokine CXCL2 in Mice. Cells, 9(10), 2296. https://doi.org/10.3390/cells9102296