Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. RNA Expression and Real-Time PCR
2.3. Genotyping Assays for Primary Cells
2.4. Lipid Droplet Staining of Primary Hepatocytes
2.5. Human Primary Hepatocyte Cell Culture
2.6. Primary Human Hepatic Stellate Cell Culture
2.7. Spheroid Cell Lines
2.8. 3D Spheroid Culture
2.9. Induction of Steatosis
2.10. Drug Treatments for Spheroids
2.11. Lipid Assay in Spheroids
2.12. Oil Red O Staining
2.13. Chromatin Immunoprecipitation (ChIP) Assays
2.14. Assay of Transposase-Accessible Chromatin (ATAC) Assays
2.15. Statistical Analysis
3. Results
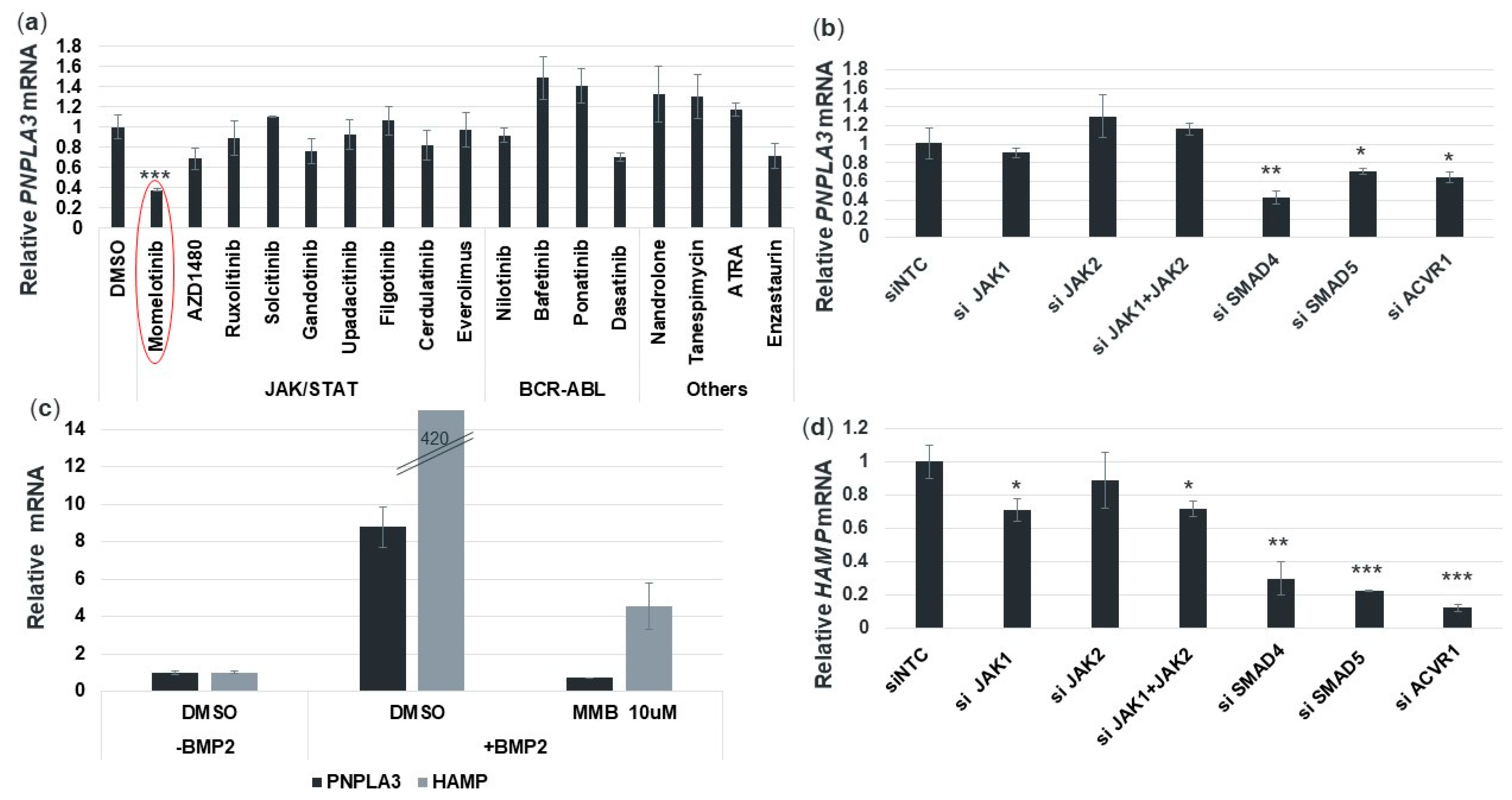
3.1. Pathway-Tailored Perturbations Identify Momelotinib as a Potential Regulator of PNPLA3 Expression
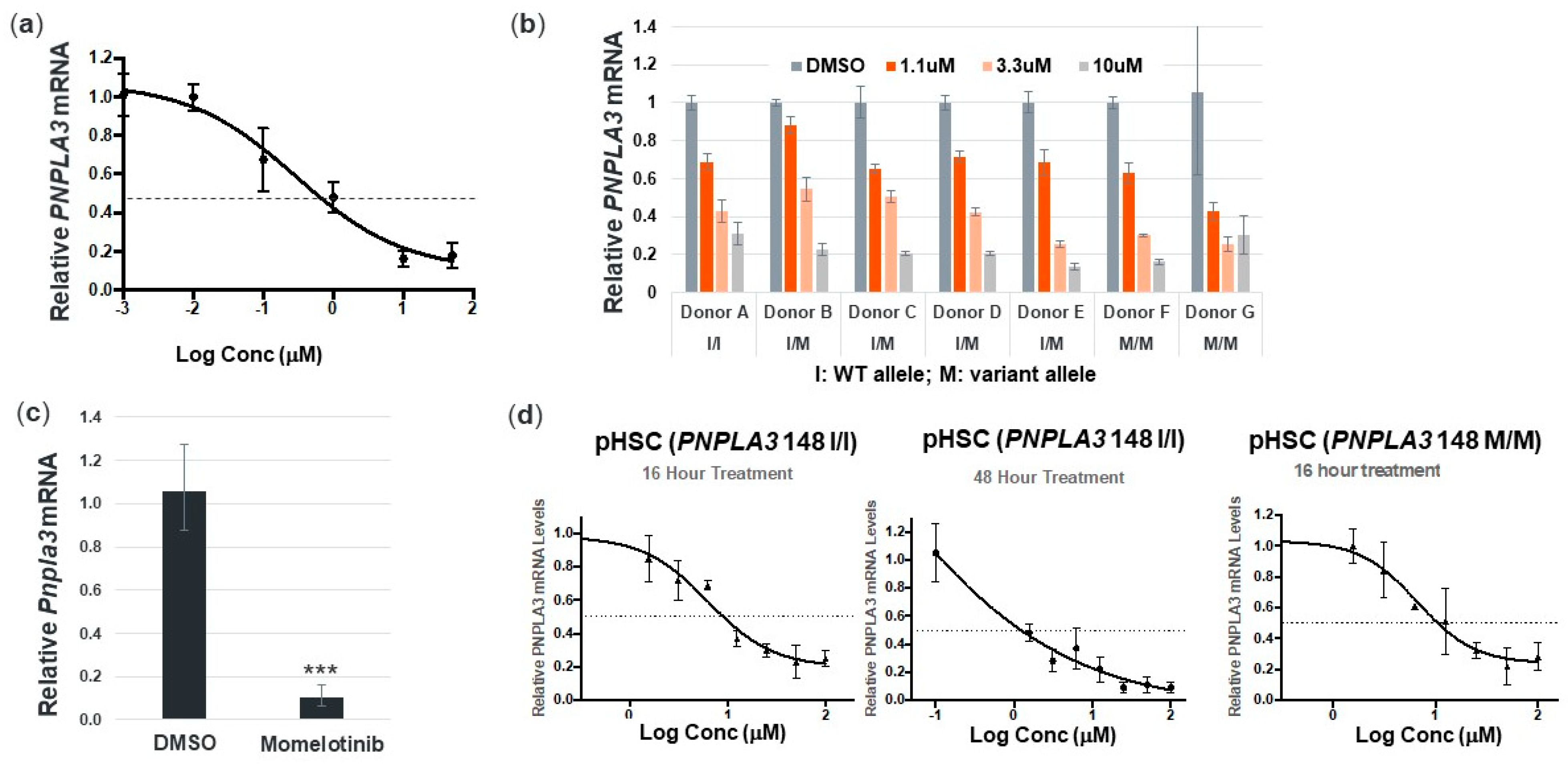
3.2. Momelotinib Reduces PNPLA3 in Human Hepatocytes and Stellate Cells from Multiple Donors, as well as Mouse Hepatocytes
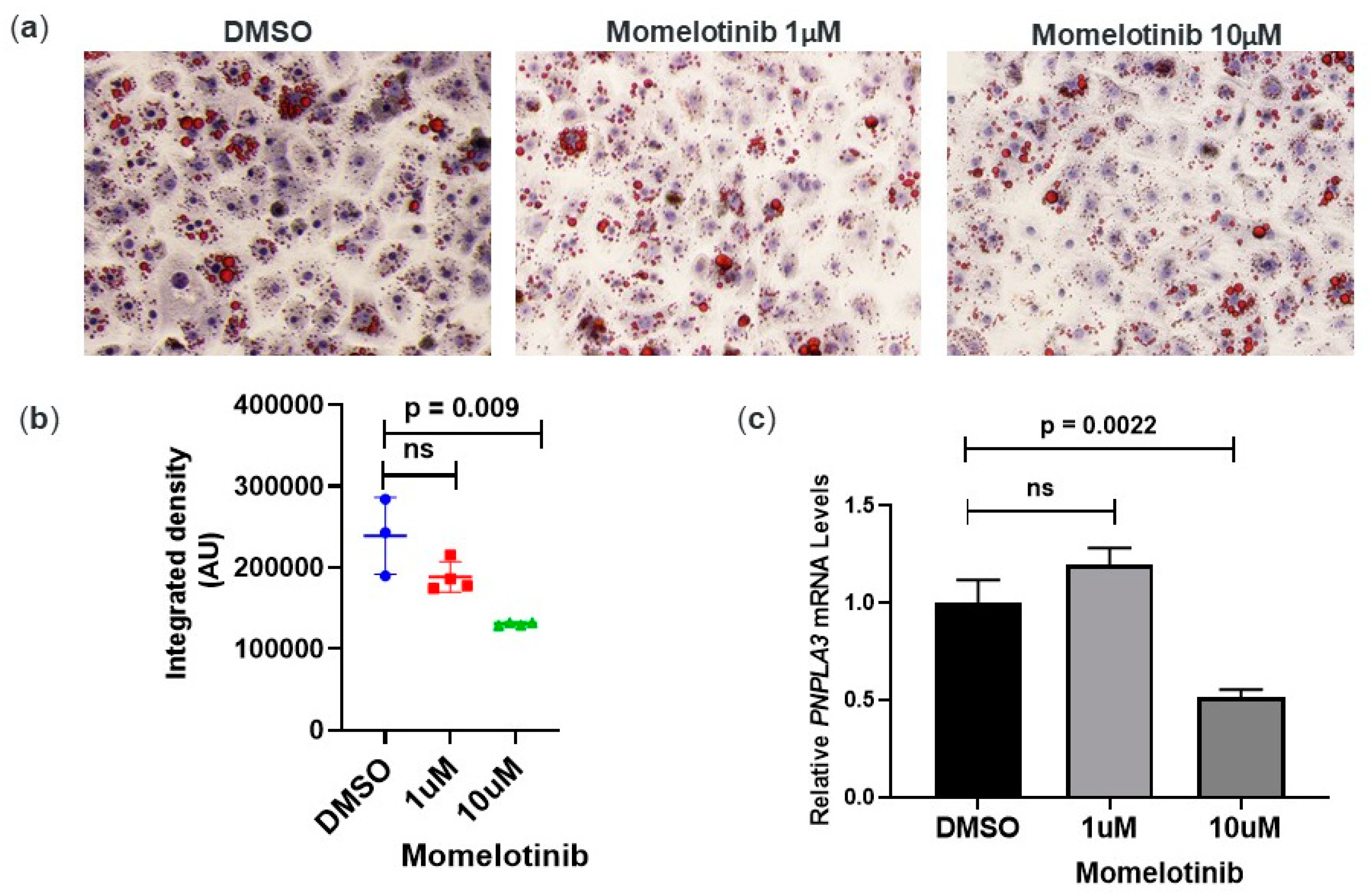
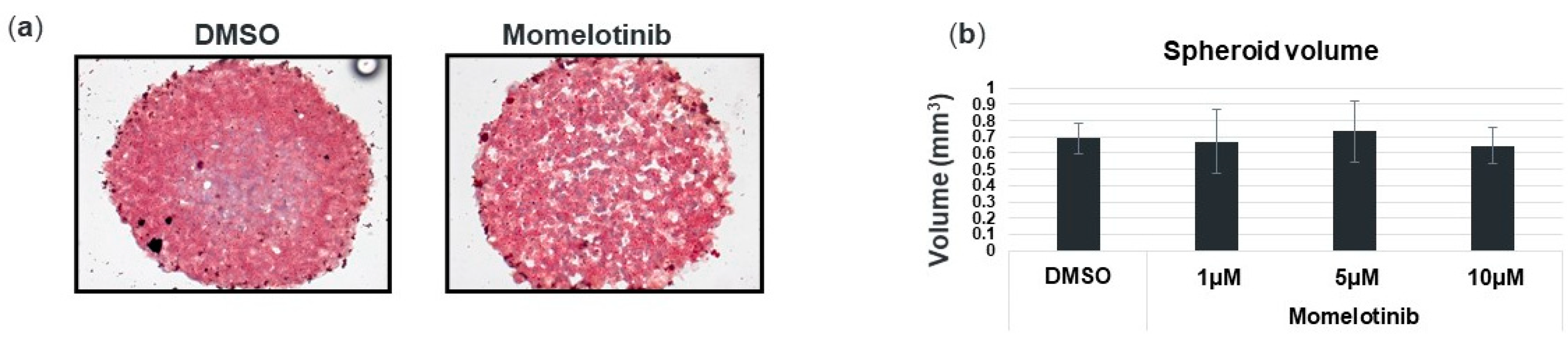
3.3. Momelotinib Reduces Lipid Droplet Content In Vitro
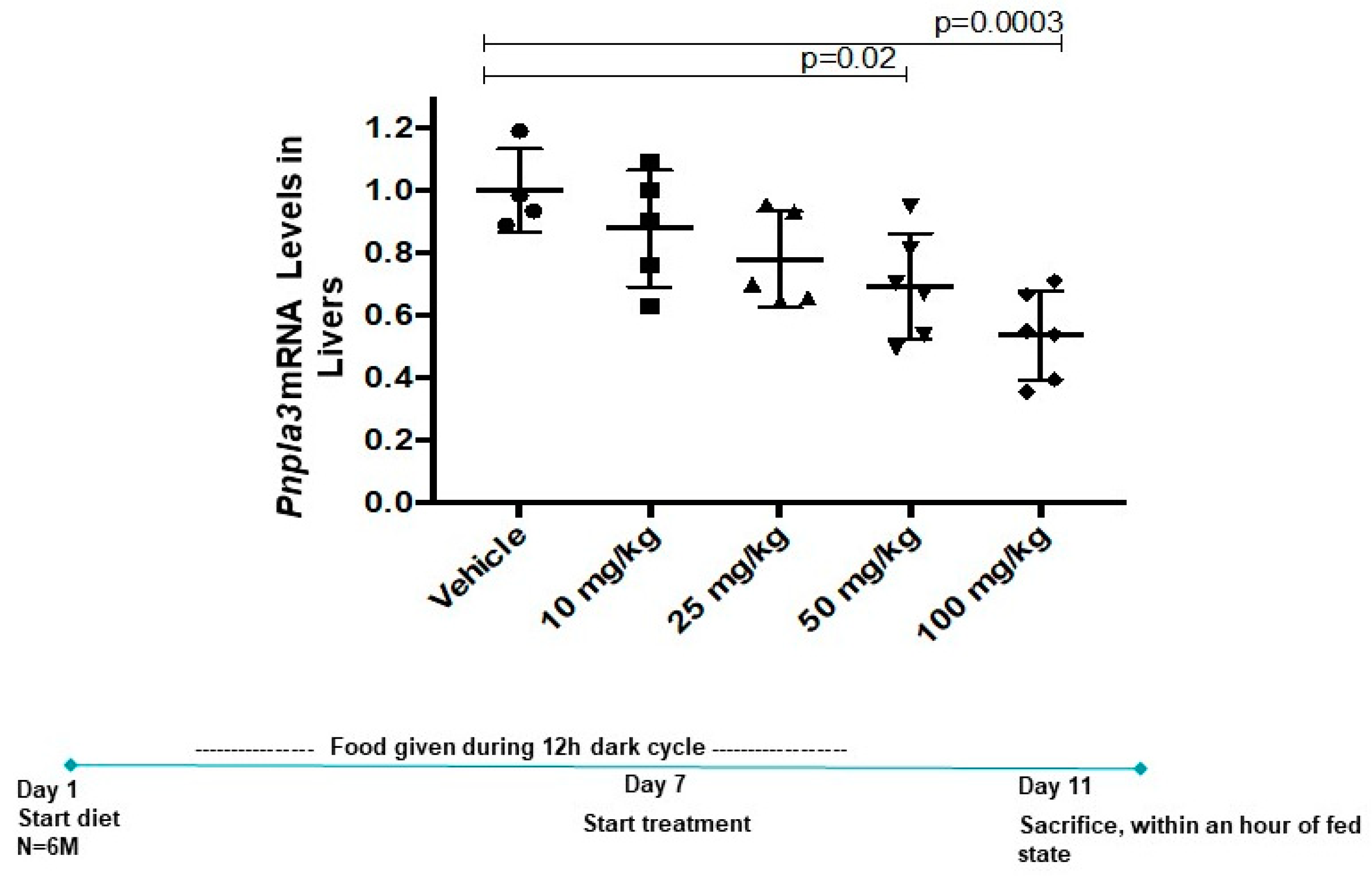
3.4. Momelotinib Reduces PNPLA3 mRNA In Vivo
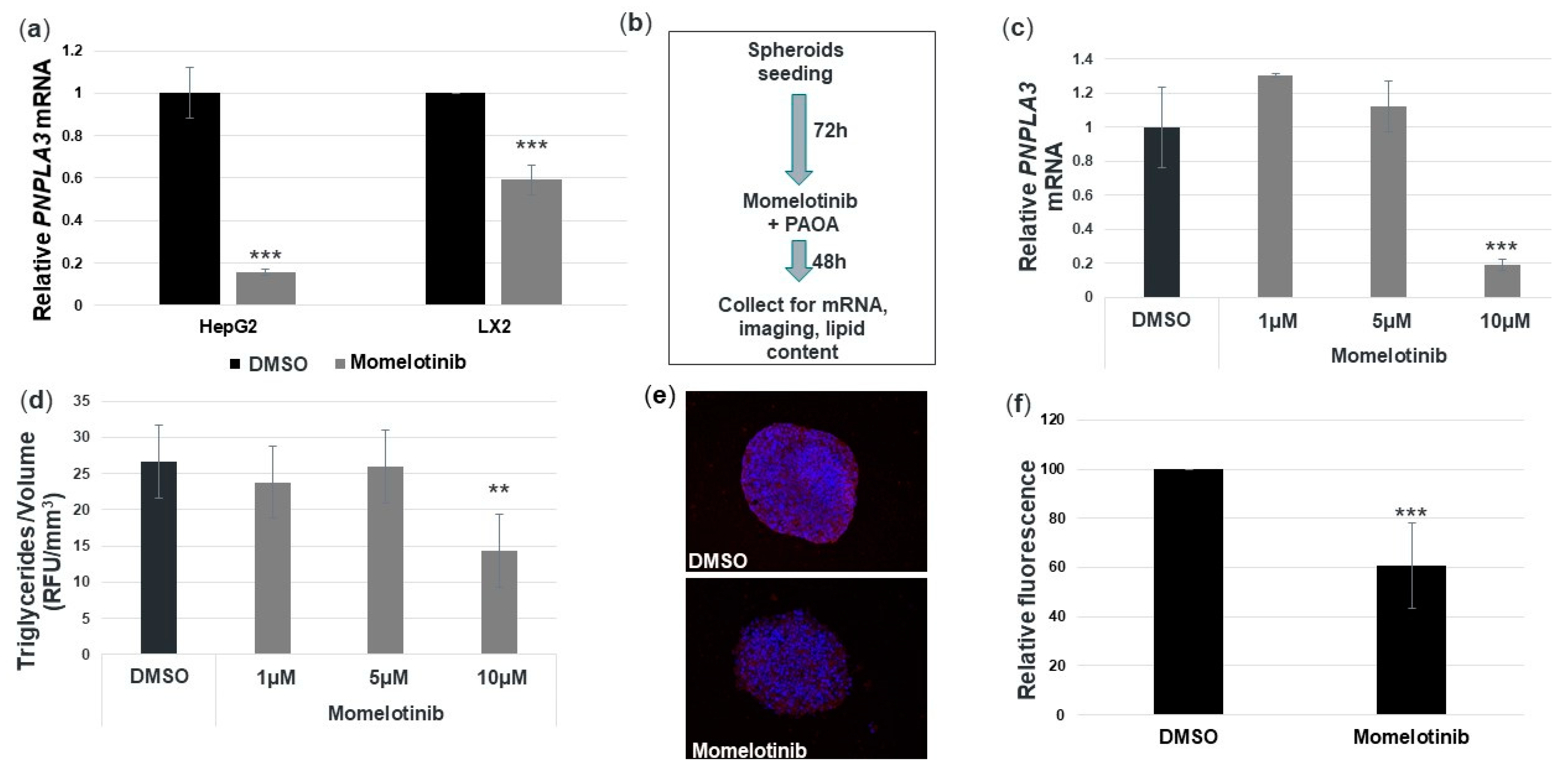
3.5. Momelotinib Reduces PNPLA3 mRNA and Triglycerides in a Novel In Vitro NASH Model
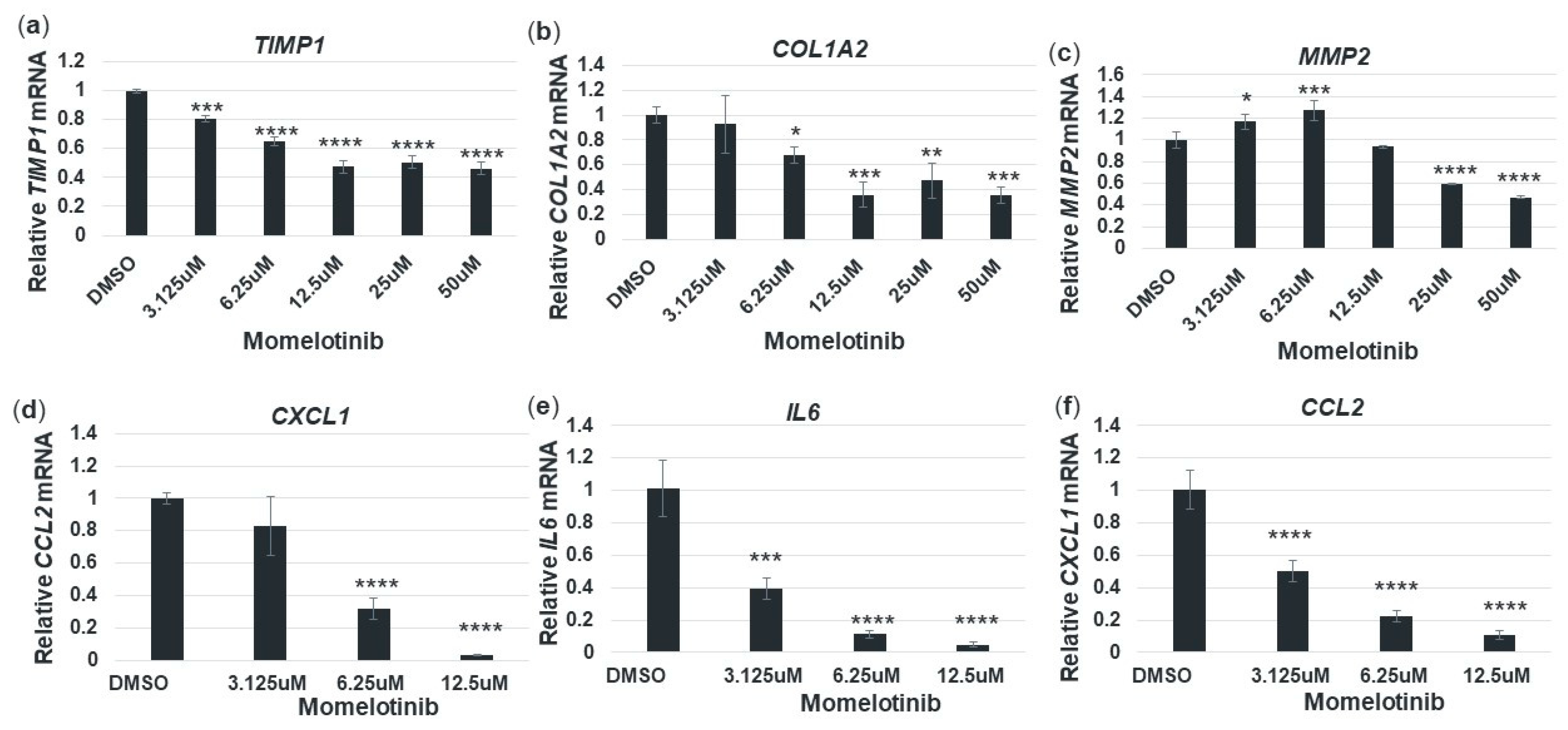
3.6. Momelotinib Reduces Expression of Profibrotic and Proinflammatory Genes in Primary Human Hepatic Stellate Cells
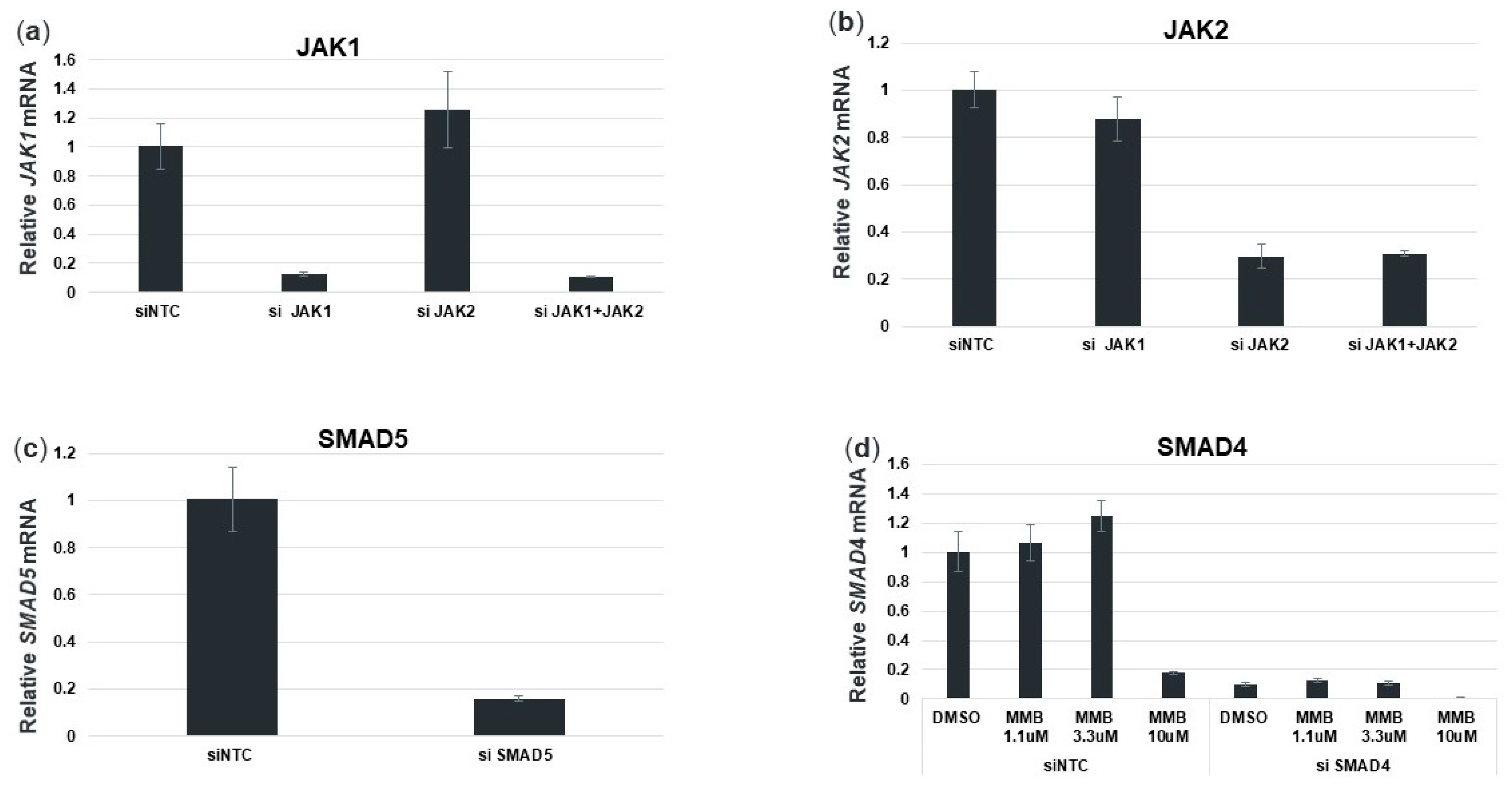
3.7. Momelotinib Reduces PNPLA3 Expression via Inhibition of the BMP Signaling Pathway
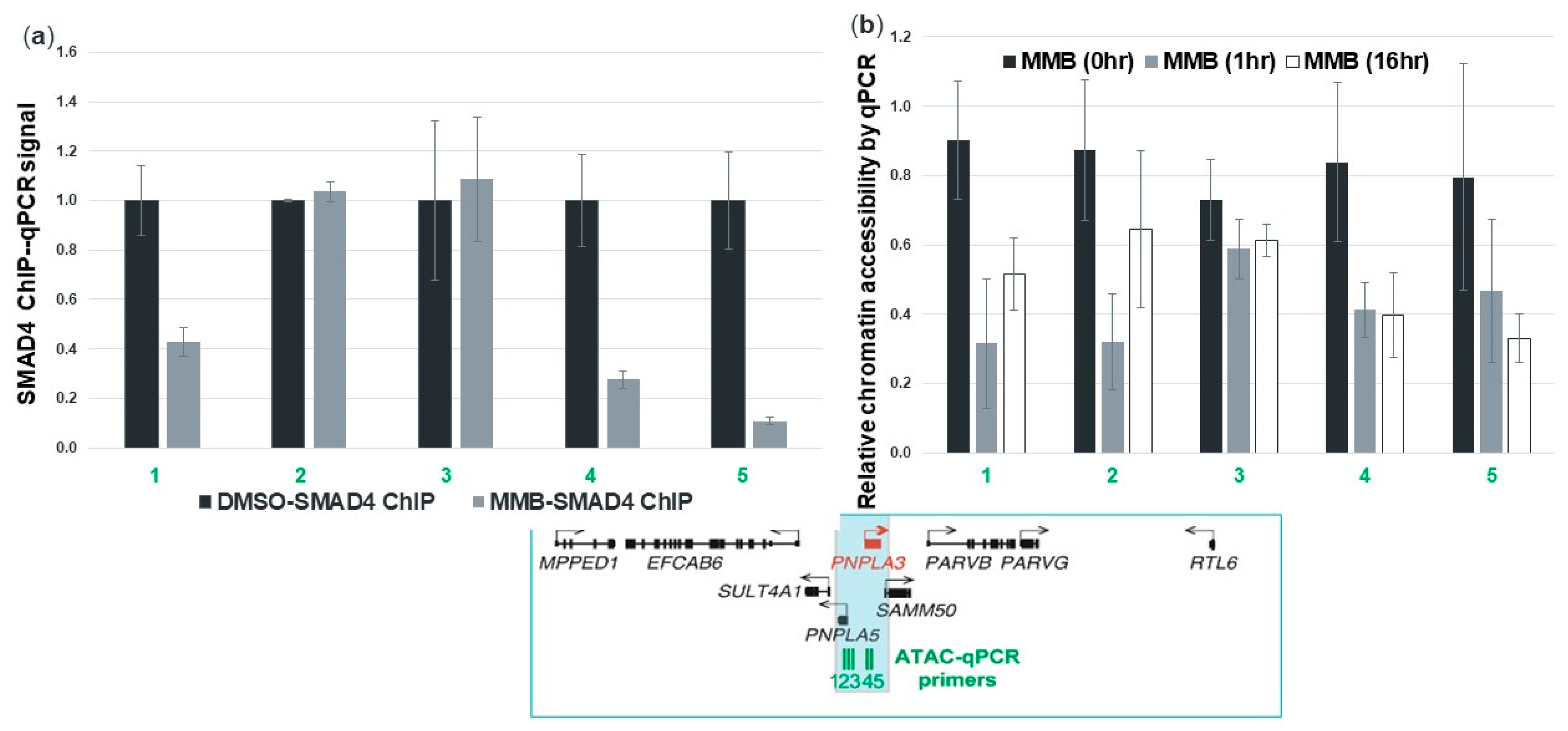
3.8. Momelotinib Reduces Chromatin Accessibility at the PNPLA3 Gene Locus
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Jakobsdottir, J.; Gorin, M.B.; Conley, Y.P.; Ferrell, R.E.; Weeks, D.E. Interpretation of genetic association studies: Markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet. 2009, 5, e1000337. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A. Bringing genome-wide association findings into clinical use. Nat. Rev. Genet. 2013, 14, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C > G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andreasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.; Smith, G.; Burns, C.J.; Fantino, E.; Tefferi, A. CYT387, a selective JAK1/JAK2 inhibitor: In vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia 2009, 23, 1441–1445. [Google Scholar] [CrossRef]
- Tyner, J.W.; Bumm, T.G.; Deininger, J.; Wood, L.; Aichberger, K.J.; Loriaux, M.M.; Druker, B.J.; Burns, C.J.; Fantino, E.; Deininger, M.W. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010, 115, 5232–5240. [Google Scholar] [CrossRef]
- Asshoff, M.; Petzer, V.; Warr, M.R.; Haschka, D.; Tymoszuk, P.; Demetz, E.; Seifert, M.; Posch, W.; Nairz, M.; Maciejewski, P.; et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 2017, 129, 1823–1830. [Google Scholar] [CrossRef]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Linden, D.; Romeo, S. Human multilineage 3D spheroids as a model of liver steatosis and fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef]
- Nicoletti, A.; Kaveri, S.; Caligiuri, G.; Bariéty, J.; Hansson, G.K. Immunoglobulin treatment reduces atherosclerosis in apo E knockout mice. J. Clin. Investig. 1998, 102, 910–918. [Google Scholar] [CrossRef]
- Lee, T.I.; Johnstone, S.E.; Young, R.A. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 2006, 1, 729–748. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Ng, K.; Hendifar, A.; Starodub, A.; Chaves, J.; Yang, Y.; Koh, B.; Barbie, D.; Hahn, W.C.; Fuchs, C.S. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2019, 37, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Yoshimura, T.; Cantley, J.L.; Majumdar, S.K.; Guebre-Egziabher, F.; Kursawe, R.; Vatner, D.F.; Fat, I.; Kahn, M.; Erion, D.M.; et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 2013, 57, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.; Mueller, T.D. Specification of BMP signaling. Cells 2019, 8, 1579. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Castano, G.O.; Burgueno, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid Res. 2009, 50, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Butler, J.L.; Palmer, C.D.; Voight, B.F.; Consortium, G.; Consortium, M.I.; Nash, C.R.N.; Hirschhorn, J.N. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010, 52, 904–912. [Google Scholar] [CrossRef]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J.; Nash, C.R.N. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwartz, B.E.; Rajagopal, V.; Smith, C.; Cohick, E.; Whissell, G.; Gamboa, M.; Pai, R.; Sigova, A.; Grossman, I.; Bumcrot, D.; et al. Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells 2020, 9, 2247. https://doi.org/10.3390/cells9102247
Schwartz BE, Rajagopal V, Smith C, Cohick E, Whissell G, Gamboa M, Pai R, Sigova A, Grossman I, Bumcrot D, et al. Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells. 2020; 9(10):2247. https://doi.org/10.3390/cells9102247
Chicago/Turabian StyleSchwartz, Brian E., Vaishnavi Rajagopal, Cynthia Smith, Evan Cohick, Gavin Whissell, Mario Gamboa, Rutuja Pai, Alla Sigova, Iris Grossman, David Bumcrot, and et al. 2020. "Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings" Cells 9, no. 10: 2247. https://doi.org/10.3390/cells9102247
APA StyleSchwartz, B. E., Rajagopal, V., Smith, C., Cohick, E., Whissell, G., Gamboa, M., Pai, R., Sigova, A., Grossman, I., Bumcrot, D., Sasidharan, K., Romeo, S., Sehgal, A., & Pingitore, P. (2020). Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells, 9(10), 2247. https://doi.org/10.3390/cells9102247