Playing Jekyll and Hyde—The Dual Role of Lipids in Fatty Liver Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Lipids and Liver Diseases
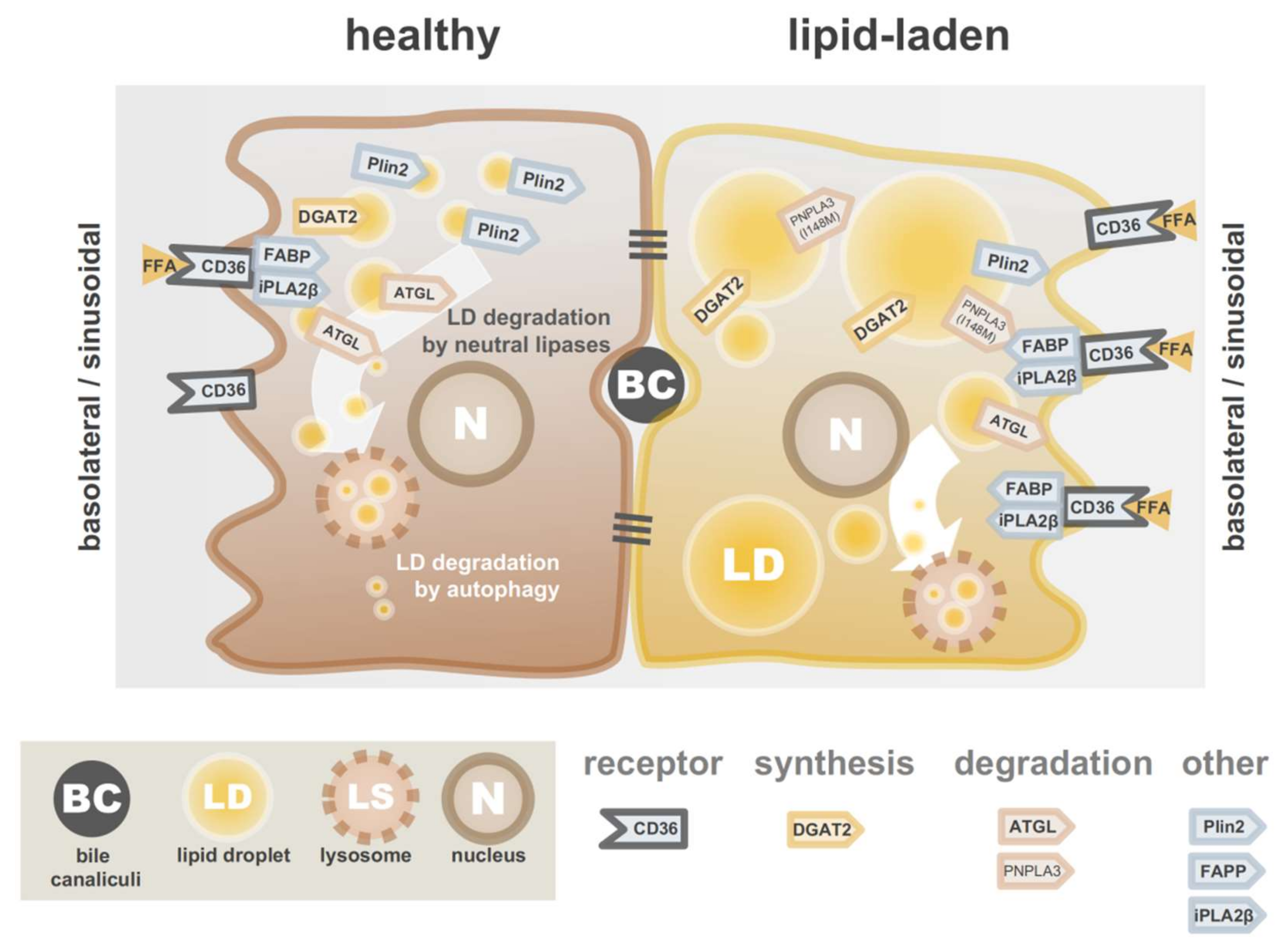
3. Lipid Accumulation in Hepatocytes
3.1. Limiting Overload of Lipids from Exogenous Sources
3.2. Limiting Endogenous Overproduction of Lipids
3.3. Promoting Lipid Catabolism and Lipid Secretion
3.4. A Reduction in Lipid Accumulation by Other Mechanisms
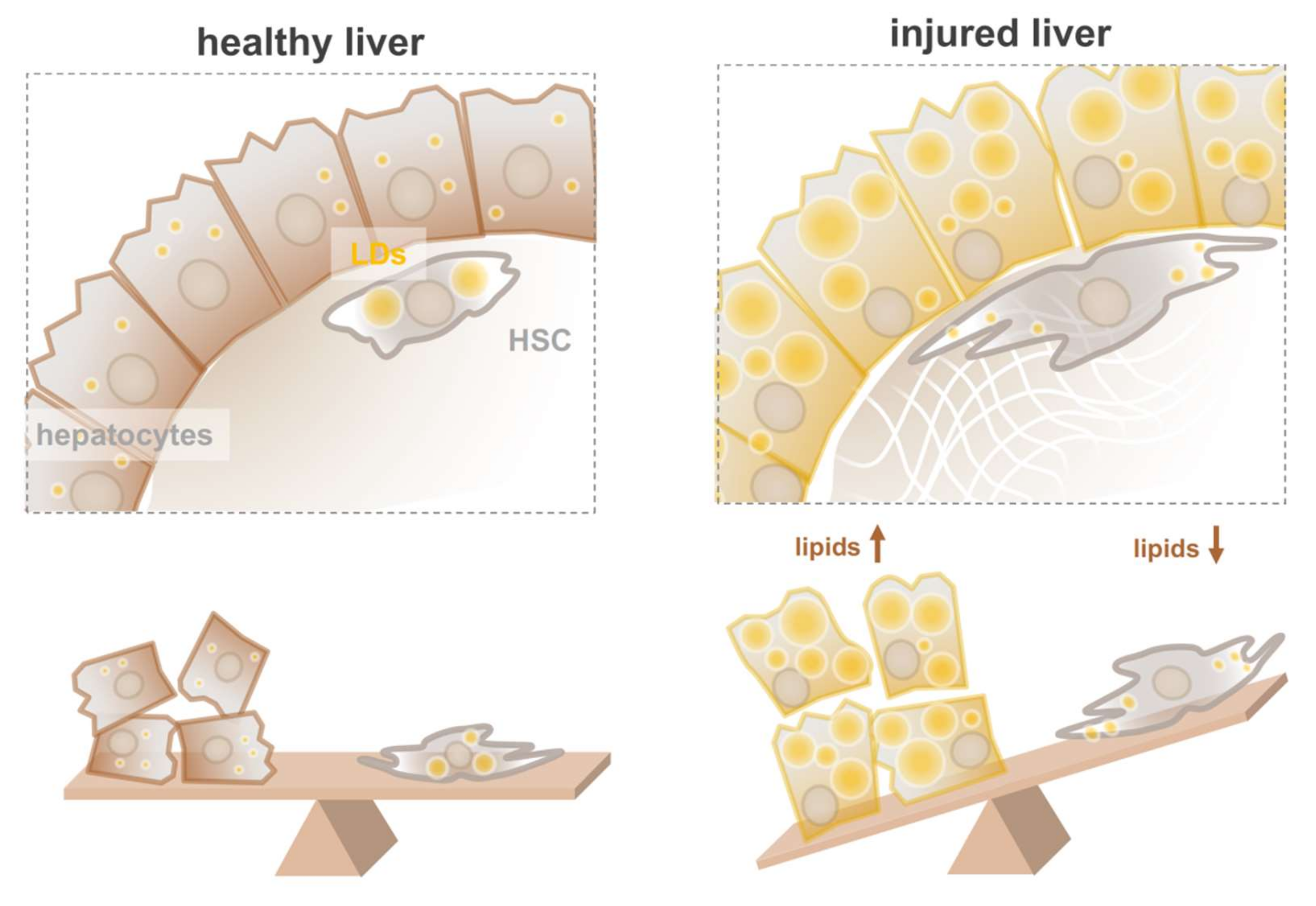
4. Hepatic Stellate Cells in Healthy and Diseased Liver
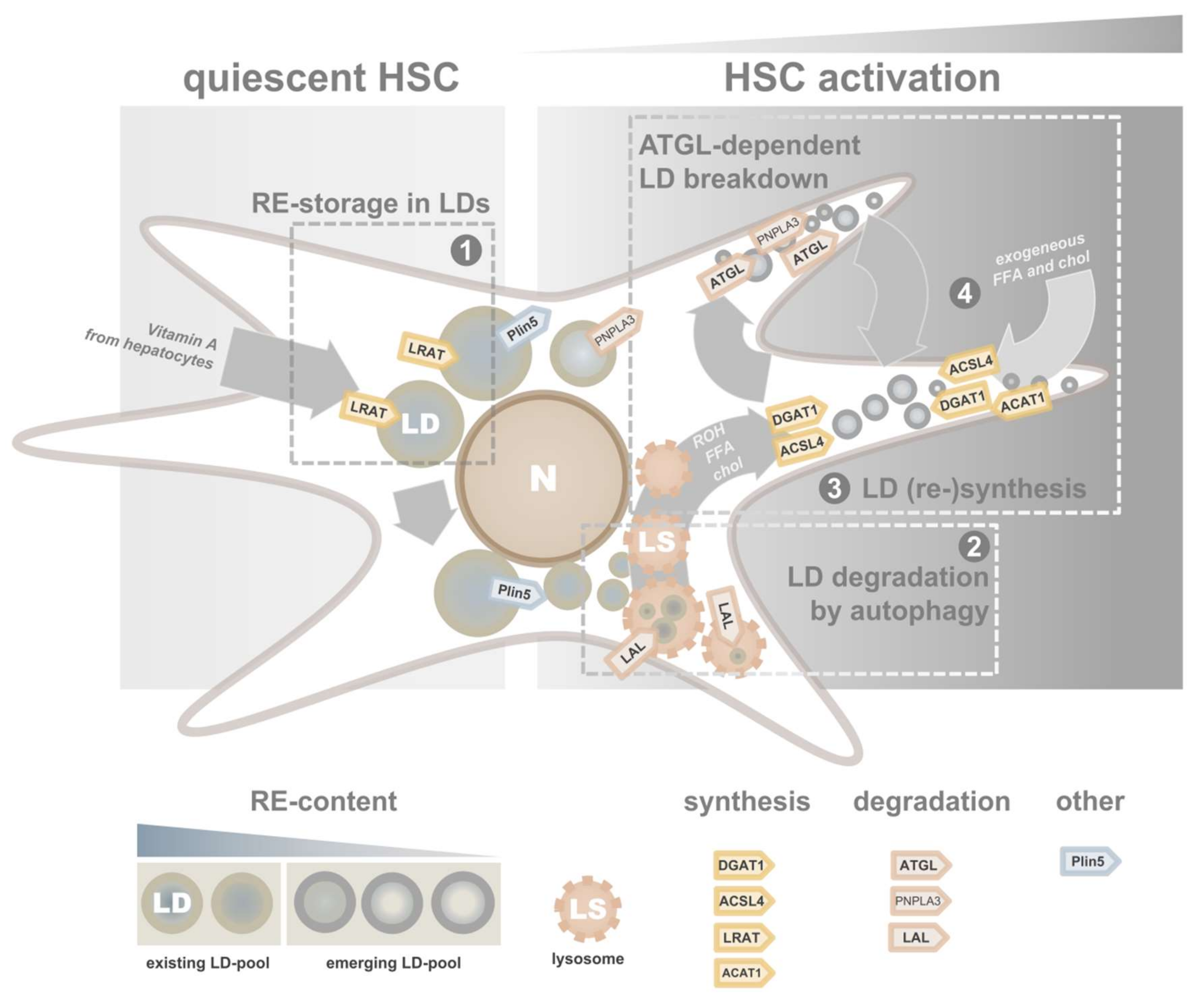
4.1. Lipid Droplet Dynamics in Activating HSCs
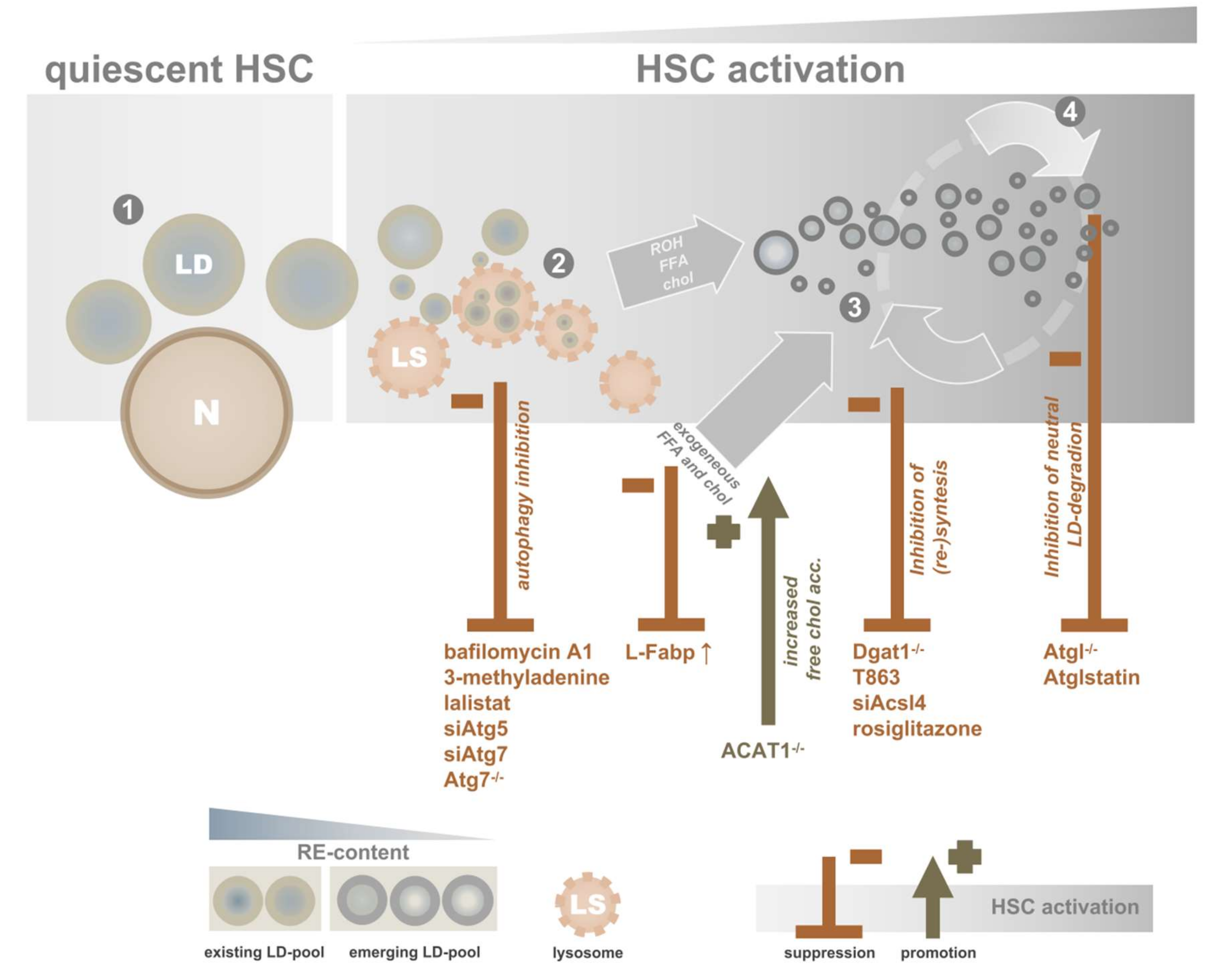
4.2. Interfering by Targeting Specific LDs
4.2.1. Targeting the “Original” LD Pool
4.2.2. Targeting the “New” LD Pool
4.2.3. Targeting Cholesterol(esters)
5. Advanced In Vitro Models for Studying Liver Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Brügger, B. Lipidomics: Analysis of the Lipid Composition of Cells and Subcellular Organelles by Electrospray Ionization Mass Spectrometry. Annu. Rev. Biochem. 2014, 83, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.B.; Kaloyanova, D.V.; Strating, J.R.P.M.; Van Hellemond, J.; Van Der Schaar, H.M.; Tielens, A.G.M.; Van Kuppeveld, F.J.M.; Brouwers, J.F. Targeting of the Hydrophobic Metabolome by Pathogens. Traffic 2015, 16, 439–460. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef]
- Thiam, A.R.; Beller, M. The why, when and how of lipid droplet diversity. J. Cell Sci. 2017, 130, 315–324. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Egan, J.J.; A Wek, S.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar]
- Jiang, H.P.; Serrero, G. Isolation and characterization of a full-length cDNA coding for an adipose differentiation-related protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7856–7860. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Barber, T.; E Wolins, N.; Serrero, G.; Blanchette-Mackie, E.J.; Londos, C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J. Lipid Res. 1997, 38, 2249–2263. [Google Scholar] [PubMed]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, K.M.; Binns, D.; Bartz, R.; Grishin, N.V.; Li, W.-P.; Agarwal, A.K.; Garg, A.; Anderson, R.G.W.; Goodman, J.M. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. USA 2007, 104, 20890–20895. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Arlt, H.; Brock, K.P.; Lai, Z.W.; DiMaio, F.; Marks, D.S.; Liao, M.; Farese, R.V.; Walther, T.C. Cryo–electron microscopy structure of the lipid droplet–formation protein seipin. J. Cell Biol. 2018, 217, 4080–4091. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Wu, X.; Lambert, T.J.; Lai, Z.W.; Walther, T.C.; Farese, R.V. LDAF1 and Seipin Form a Lipid Droplet Assembly Complex. Dev. Cell 2019, 51, 551–563. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Steib, C. Kupffer cell activation and portal hypertension. Gut 2011, 60, 1307–1308. [Google Scholar] [CrossRef][Green Version]
- Banales, J.-M.; Prieto, J.; Medina, J.-F. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J. Gastroenterol. 2006, 12, 3496–3511. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; She, H.; Hazra, S.; Cheng, J.; Wang, J. Fat paradox of steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, S104–S107. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Anstee, Q.M.; McPherson, S. Non-alcoholic fatty liver disease: A practical approach to diagnosis and staging. Frontline Gastroenterol. 2014, 5, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Gomez, M.R.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Coleman, I.S.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005, 115, 1343. [Google Scholar] [CrossRef]
- Brunt, E.M.; Neuschwander-Tetri, B.A.; Oliver, D.; Wehmeier, K.R.; Bacon, B.R. Nonalcoholic steatohepatitis: Histologic features and clinical correlations with 30 blinded biopsy specimens. Hum. Pathol. 2004, 35, 1070–1082. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Ikura, Y.; Dias, D.; Isomoto, K.; Yabu, A.; Moskaluk, C.; Pramoonjago, P.; Simmons, W.; Scruggs, H.; Rosenbaum, N.; et al. Hepatocellular ballooning in NASH. J. Hepatol. 2010, 53, 719–723. [Google Scholar] [CrossRef]
- Chitraju, C.; Trötzmüller, M.; Hartler, J.; Wolinski, H.; Thallinger, G.G.; Lass, A.; Zechner, R.; Zimmermann, R.; Köfeler, H.; Spener, F. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J. Lipid Res. 2012, 53, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Nergiz-Unal, R.; Rademakers, T.; Cosemans, J.M.; Heemskerk, J.W. CD36 as a multiple-ligand signaling receptor in atherothrombosis. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Stremmel, W.; Strohmeyer, G.; Berk, P.D. Hepatocellular uptake of oleate is energy dependent, sodium linked, and inhibited by an antibody to a hepatocyte plasma membrane fatty acid binding protein. Proc. Natl. Acad. Sci. USA 1986, 83, 3584–3588. [Google Scholar] [CrossRef]
- Stremmel, W.; Strohmeyer, G.; Borchard, F.; Kochwa, S.; Berk, P.D. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc. Natl. Acad. Sci. USA 1985, 82, 4–8. [Google Scholar] [CrossRef]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Wang, F.-S.; Yang, Y.-L.; Huang, Y.-H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Kuo, H.-C.; Wang, F.-S.; Huang, Y.-H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Itami, S.; Kuroda, M.; Yoshizato, K.; Kawada, N.; Murakami, Y. MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice. Mol. Ther. 2016, 24, 1848–1859. [Google Scholar] [CrossRef]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.-X.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Staffer, S.; Wannhoff, A.; Pathil, A.; Chamulitrat, W. Plasma membrane phospholipase A 2 controls hepatocellular fatty acid uptake and is responsive to pharmacological modulation: Implications for nonalcoholic steatohepatitis. FASEB J. 2014, 28, 3159–3170. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Senturk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Tontonoz, P.; Mangelsdorf, D.J. Liver X Receptor Signaling Pathways in Cardiovascular Disease. Mol. Endocrinol. 2003, 17, 985–993. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V. Thematic Review Series: Glycerolipids.DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar]
- Zammit, V. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem. J. 2013, 451, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Wurie, H.R.; Buckett, L.; Zammit, V. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS J. 2012, 279, 3033–3047. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J. Lipid Res. 2012, 53, 1106–1116. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- McLaren, D.G.; Han, S.; Murphy, B.A.; Wilsie, L.; Stout, S.J.; Zhou, H.; Roddy, T.P.; Gorski, J.N.; Metzger, D.E.; Shin, M.K.; et al. DGAT2 Inhibition Alters Aspects of Triglyceride Metabolism in Rodents but Not in Non-human Primates. Cell Metab. 2018, 27, 1236–1248.e6. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Gabriel, K.R.; Chitraju, C.; Bronson, R.T.; Mejhert, N.; Boland, S.; Wang, K.; Lai, Z.W.; Farese, R.V., Jr.; Walther, T.C. Hepatocyte Deletion of Triglyceride-Synthesis Enzyme Acyl CoA: Diacylglycerol Acyltransferase 2 Reduces Steatosis Without Increasing Inflammation or Fibrosis in Mice. Hepatology 2019, 70, 1972–1985. [Google Scholar] [CrossRef]
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; A Hannan, L.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: A multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838. [Google Scholar] [CrossRef]
- Amin, N.B.; Carvajal-Gonzalez, S.; Purkal, J.; Zhu, T.; Crowley, C.; Perez, S.; Chidsey, K.; Kim, A.M.; Goodwin, B. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci. Transl. Med. 2019, 11, eaav9701. [Google Scholar] [CrossRef]
- Xiao, W.; Ren, M.; Zhang, C.; Li, S.; An, W. Amelioration of nonalcoholic fatty liver disease by hepatic stimulator substance via preservation of carnitine palmitoyl transferase-1 activity. Am. J. Physiol. Cell Physiol. 2015, 309, C215–C227. [Google Scholar] [CrossRef]
- Tobita, H.; Sato, S.; Yazaki, T.; Mishiro, T.; Ishimura, N.; Ishihara, S.; Kinoshita, Y. Alogliptin alleviates hepatic steatosis in a mouse model of nonalcoholic fatty liver disease by promoting CPT1a expression via Thr172 phosphorylation of AMPKα in the liver. Mol. Med. Rep. 2018, 17, 6840–6846. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Meza-Rios, A.; Garcia-Bañuelos, J.; Vera-Cruz, J.; Gutiérrez-Cuevas, J.; Silva-Gomez, J.; Staels, B.; Dominguez-Rosales, J.; Galicia-Moreno, M.; et al. Pirfenidone Is an Agonistic Ligand for PPARα and Improves NASH by Activation of SIRT1/LKB1/pAMPK. Hepatol. Commun. 2020, 4, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Fukuo, Y.; Yamashina, S.; Sonoue, H.; Arakawa, A.; Nakadera, E.; Aoyama, T.; Uchiyama, A.; Kon, K.; Ikejima, K.; Watanabe, S. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol. Res. 2014, 44, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Kashima, J.; Shintani-Ishida, K.; Nakajima, M.; Maeda, H.; Unuma, K.; Uchiyama, Y.; Yoshida, K.-I. Immunohistochemical study of the autophagy marker microtubule-associated protein 1 light chain 3 in normal and steatotic human livers. Hepatol. Res. 2013, 44, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; Panayotova-Dimitrova, D.; Leverkus, M.; Vondran, F.W.; Bauerfeind, R.; Binz, A.; Behrens, G.M. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS 2012, 26, 1995–2006. [Google Scholar] [CrossRef]
- Lin, C.-W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.-M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef]
- Park, H.-W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.-H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.-J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Najt, C.P.; Senthivinayagam, S.; Aljazi, M.B.; Fader, K.A.; Olenic, S.D.; Brock, J.R.L.; Lydic, T.A.; Jones, A.D.; Atshaves, B.P. Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am. J. Physiol. Liver Physiol. 2016, 310, G726–G738. [Google Scholar] [CrossRef] [PubMed]
- Libby, A.E.; Bales, E.; Orlicky, D.J.; McManaman, J.L. Perilipin-2 Deletion Impairs Hepatic Lipid Accumulation by Interfering with Sterol Regulatory Element-binding Protein (SREBP) Activation and Altering the Hepatic Lipidome. J. Biol. Chem. 2016, 291, 24231–24246. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.; Townsend, E.C.; Rickertsen, C.; Hains, A.; Brown, E.; Inwards, E.G.; Stoeckman, A.K.; Matis, M.P.; Sampathkumar, R.S.; Osna, N.A.; et al. Decreasing Phosphatidylcholine on the Surface of the Lipid Droplet Correlates with Altered Protein Binding and Steatosis. Cells 2018, 7, 230. [Google Scholar] [CrossRef] [PubMed]
- Orlicky, D.J.; Libby, A.E.; Bales, E.S.; Mcmahan, R.H.; Monks, J.; La Rosa, F.G.; McManaman, J.L. Perilipin-2 promotes obesity and progressive fatty liver disease in mice through mechanistically distinct hepatocyte and extra-hepatocyte actions. J. Physiol. 2019, 597, 1565–1584. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ojha, S.; Rai, P.; Joshi, A.; Kamat, S.S.; Mallik, R. Insulin activates intracellular transport of lipid droplets to release triglycerides from the liver. J. Cell Biol. 2019, 218, 3697–3713. [Google Scholar] [CrossRef]
- Czech, M.P.; Tencerova, M.; Pedersen, D.J.; Aouadi, M. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 2013, 56, 949–964. [Google Scholar] [CrossRef]
- Ye, J.; Li, J.Z.; Liu, Y.; Li, X.; Yang, T.; Ma, X.; Li, Q.; Yao, Z.; Li, P. Cideb, an ER- and Lipid Droplet-Associated Protein, Mediates VLDL Lipidation and Maturation by Interacting with Apolipoprotein B. Cell Metab. 2009, 9, 177–190. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in Adipose Tissue and Hepatic Lipid Kinetics in Obese Men and Women With Nonalcoholic Fatty Liver Disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimano, H.; Hamilton, R.L.; Brown, M.S.; Goldstein, J.L. Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J. Clin. Investig. 1999, 103, 1067–1076. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Yuan, X.; Waterworth, D.; Perry, J.R.; Lim, N.; Song, K.; Chambers, J.C.; Zhang, W.; Vollenweider, P.; Stirnadel, H.; Johnson, T.; et al. Population-Based Genome-wide Association Studies Reveal Six Loci Influencing Plasma Levels of Liver Enzymes. Am. J. Hum. Genet. 2008, 83, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Smagris, E.; Basuray, S.; Li, J.Z.; Huang, Y.; Lai, K.-M.V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148Mknockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2014, 61, 108–118. [Google Scholar] [CrossRef]
- Basuray, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef]
- Basuray, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef]
- Kory, N.; Thiam, A.-R.; Farese, R.V.; Walther, T.C. Protein Crowding Is a Determinant of Lipid Droplet Protein Composition. Dev. Cell 2015, 34, 351–363. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Dong, X.C. PNPLA3—A Potential Therapeutic Target for Personalized Treatment of Chronic Liver Disease. Front. Med. 2019, 6, 304. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Tóth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; E Moor, A.; et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 1–5. [Google Scholar] [CrossRef]
- Park, S.R.; Cho, C.-S.; Xi, J.; Kang, H.M.; Lee, J.H. Holistic Characterization of Single Hepatocyte Transcriptome Responses to High Fat Diet. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Yamada, M.; Blaner, W.S.; Soprano, D.R.; Dixon, J.L.; Kjeldbye, H.M.; Goodman, D.S. Biochemical characteristics of isolated rat liver stellate cells. Hepatology 1987, 7, 1224–1229. [Google Scholar] [CrossRef]
- Moriwaki, H.; Blaner, W.S.; Piantedosi, R.; Goodman, D.S. Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J. Lipid Res. 1988, 29, 1523–1534. [Google Scholar]
- Wake, K. Development of vitamin A-rich lipid droplets in multivesicular bodies of rat liver stellate cells. J. Cell Biol. 1974, 63, 683–691. [Google Scholar] [CrossRef]
- D’Ambrosio, D.N.; Walewski, J.L.; Clugston, R.D.; Berk, P.D.; Rippe, R.A.; Blaner, W.S. Distinct Populations of Hepatic Stellate Cells in the Mouse Liver Have Different Capacities for Retinoid and Lipid Storage. PLoS ONE 2011, 6, e24993. [Google Scholar] [CrossRef]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; Van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.J.; Lee, S.-A.; Jiang, H.; Brun, P.-J.; Blaner, W.S. DGAT1-deficiency affects the cellular distribution of hepatic retinoid and attenuates the progression of CCl4-induced liver fibrosis. Hepatobiliary Surg. Nutr. 2015, 4, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Hundertmark, J.; Ritz, T.P.; Weiskirchen, R.; Tacke, F. Single Cell RNA Sequencing Identifies Subsets of Hepatic Stellate Cells and Myofibroblasts in Liver Fibrosis. Cells 2019, 8, 503. [Google Scholar] [CrossRef]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847.e8. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; Van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol Acyltransferase Is Essential for Accumulation of All-trans-Retinyl Esters in the Eye and in the Liver. J. Biol. Chem. 2003, 279, 10422–10432. [Google Scholar] [CrossRef]
- Liu, L.; Gudas, L.J. Disruption of the Lecithin:Retinol Acyltransferase Gene Makes Mice More Susceptible to Vitamin A Deficiency. J. Biol. Chem. 2005, 280, 40226–40234. [Google Scholar] [CrossRef]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.J.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid Absorption and Storage Is Impaired in Mice Lacking Lecithin:Retinol Acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar] [CrossRef]
- Wongsiriroj, N.; Piantedosi, R.; Palczewski, K.; Goldberg, I.J.; Johnston, T.P.; Li, E.; Blaner, W.S. The Molecular Basis of Retinoid Absorption. J. Biol. Chem. 2008, 283, 13510–13519. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ogawa, K. Fine structure and cytochemistry of lysosomes in the Ito cells of the rat liver. Cell Tissue Res. 1983, 233, 45–57. [Google Scholar] [CrossRef]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.-Y.; Dapito, D.H.; Pradère, J.-P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef]
- Ajat, M.; Molenaar, M.; Brouwers, J.F.; Vaandrager, A.B.; Houweling, M.; Helms, J.B. Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.L.; Bloomer, S.A.; Chan, E.P.; Gaça, M.D.A.A.; Georges, P.C.; Sackey, B.; Uemura, M.; Janmey, P.A.; Wells, R.G. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G110–G118. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Trasino, S.E.; Tang, X.-H.; Jessurun, J.; Gudas, L.J. Obesity Leads to Tissue, but not Serum Vitamin A Deficiency. Sci. Rep. 2015, 5, 15893. [Google Scholar] [CrossRef]
- Nepomnyashchikh, G.I.; Aidagulova, S.V.; Nepomnyashchikh, D.L.; Kapustina, V.I.; Postnikova, O.A. Ultrastructural and immunohistochemical study of hepatic stellate cells over the course of infectious viral fibrosis and cirrhosis of the liver. Bull. Exp. Biol. Med. 2006, 142, 723–728. [Google Scholar] [CrossRef]
- Mansy, S.; ElKhafif, N.; AbelFatah, A.S.; Yehia, H.A.; Mostafa, I. Hepatic Stellate Cells and Fibrogenesis in Hepatitis C Virus Infection: An Ultrastructural Insight. Ultrastruct. Pathol. 2010, 34, 62–67. [Google Scholar] [CrossRef]
- Geerts, A.; Bouwens, L.; Wisse, E. Ultrastructure and function of hepatic fat-storing and pit cells. J. Electron Microsc. Tech. 1990, 14, 247–256. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Wassenaar, T.A.; Yadav, K.K.; Toulmay, A.; Mari, M.C.; Caillon, L.; Chorlay, A.; Haaker, M.W.; Wubbolts, R.W.; Houweling, M.; et al. Lecithin:Retinol Acyl Transferase (LRAT) induces the formation of lipid droplets. bioRxiv 2019, 733931. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Vaandrager, A.B.; Helms, J.B. Some Lipid Droplets Are More Equal Than Others: Different Metabolic Lipid Droplet Pools in Hepatic Stellate Cells. Lipid Insights 2017, 10, 1178635317747281. [Google Scholar] [CrossRef]
- Senoo, H.; Wake, K. Suppression of experimental hepatic fibrosis by administration of vitamin A. Lab. Invest. 1985, 52, 182–194. [Google Scholar]
- Davis, B.H.; Kramer, R.T.; O Davidson, N. Retinoic acid modulates rat Ito cell proliferation, collagen, and transforming growth factor beta production. J. Clin. Investig. 1990, 86, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.H.; Coll, D.; A Beno, D.W. Retinoic acid suppresses the response to platelet-derived growth factor in human hepatic Ito-cell-like myofibroblasts: A post-receptor mechanism independent of raf/fos/jun/egr activation. Biochem. J. 1993, 294, 785–791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seifert, W.F.; Bosma, A.; Brouwer, A.; Hendriks, H.F.; Roholl, P.J.; Van Leeuwen, R.E.; Van Thiel-de Ruiter, G.C.; Seifert-Bock, I.; Knook, D.L. Vitamin A deficiency potentiates carbon tetrachloride-induced liver fibrosis in rats. Hepatology 1994, 19, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, Y.; Shimizu, I.; Yasuda, M.; Hori, H.; Shono, M.; Ito, S. Retinyl palmitate reduces hepatic fibrosis in rats induced by dimethylnitrosamine or pig serum. J. Hepatol. 1998, 29, 933–943. [Google Scholar] [CrossRef]
- Lee, T.F.; Mak, K.M.; Rackovsky, O.; Lin, Y.-L.; Kwong, A.J.; Loke, J.C.; Friedman, S.L. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J. Cell. Physiol. 2010, 223, 648–657. [Google Scholar] [CrossRef]
- Okuno, M.; Moriwaki, H.; Imai, S.; Muto, Y.; Kawada, N.; Suzuki, Y.; Kojima, S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-beta in liver stellate cells. Hepatology 1997, 26, 913–921. [Google Scholar] [CrossRef]
- Okuno, M.; Sato, T.; Kitamoto, T.; Imai, S.; Kawada, N.; Suzuki, Y.; Yoshimura, H.; Moriwaki, H.; Onuki, K.; Masushige, S.; et al. Increased 9,13-di-cis-retinoic acid in rat hepatic fibrosis: Implication for a potential link between retinoid loss and TGF-beta mediated fibrogenesis in vivo. J. Hepatol. 1999, 30, 1073–1080. [Google Scholar] [CrossRef]
- Geubel, A.P.; De Galocsy, C.; Alves, N.; Rahier, J.; Dive, C. Liver damage caused by therapeutic vitamin A administration: Estimate of dose-related toxicity in 41 cases. Gastroenterology 1991, 100, 1701–1709. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Arozena, A.A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.M.; Van Grunsven, L. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Grumet, L.; Eichmann, T.O.; Taschler, U.; Zierler, K.A.; Leopold, C.; Moustafa, T.; Radovic, B.; Romauch, M.; Yan, C.; Du, H.; et al. Lysosomal Acid Lipase Hydrolyzes Retinyl Ester and Affects Retinoid Turnover. J. Biol. Chem. 2016, 291, 17977–17987. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Molenaar, M.; Spee, B.; Brouwers, J.F.; Wubbolts, R.; Houweling, M.; Yan, C.; Du, H.; VanderVen, B.C.; Vaandrager, A.B.; et al. Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation. J. Biol. Chem. 2017, 292, 12436–12448. [Google Scholar] [CrossRef] [PubMed]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Spee, B.; Kruitwagen, H.S.H.S.; Wubbolts, R.; Brouwers, J.F.H.M.; Van de Lest, C.H.; Molenaar, M.R.; Houweling, M.; Helms, J.B.; Vaandrager, A.B. Role of long-chain acyl-CoA synthetase 4 in formation of polyunsaturated lipid species in hepatic stellate cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1851, 220–230. [Google Scholar] [CrossRef]
- Tuohetahuntila, M.; Molenaar, M.; Spee, B.; Brouwers, J.F.; Houweling, M.; Vaandrager, A.B.; Helms, J.B. ATGL and DGAT1 are involved in the turnover of newly synthesized triacylglycerols in hepatic stellate cells[S]. J. Lipid Res. 2016, 57, 1162–1174. [Google Scholar] [CrossRef]
- Mello, T.; Nakatsuka, A.; Fears, S.; Davis, W.; Tsukamoto, H.; Bosron, W.F.; Sanghani, S.P. Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun. 2008, 374, 460–464. [Google Scholar] [CrossRef]
- Taschler, U.; Schreiber, R.; Chitraju, C.; Grabner, G.F.; Romauch, M.; Wolinski, H.; Haemmerle, G.; Breinbauer, R.; Zechner, R.; Lass, A.; et al. Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 937–945. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.-P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with Adipose Triglyceride Lipase. J. Biol. Chem. 2010, 286, 5126–5135. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bell, M.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; Coleman, R.; et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef]
- Lin, J.; Chen, A. Perilipin 5 restores the formation of lipid droplets in activated hepatic stellate cells and inhibits their activation. Lab. Investig. 2016, 96, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, R.; Lee, M.-J.; Pickering, R.T.; Sharma, V.M.; Puri, V.; Kandror, K.V. Fat-specific Protein 27 Inhibits Lipolysis by Facilitating the Inhibitory Effect of Transcription Factor Egr1 on Transcription of Adipose Triglyceride Lipase. J. Biol. Chem. 2014, 289, 14481–14487. [Google Scholar] [CrossRef] [PubMed]
- Grahn, T.H.M.; Kaur, R.; Yin, J.; Schweiger, M.; Sharma, V.M.; Lee, M.-J.; Ido, Y.; Smas, C.M.; Zechner, R.; Lass, A.; et al. Fat-specific Protein 27 (FSP27) Interacts with Adipose Triglyceride Lipase (ATGL) to Regulate Lipolysis and Insulin Sensitivity in Human Adipocytes. J. Biol. Chem. 2014, 289, 12029–12039. [Google Scholar] [CrossRef]
- Yu, F.; Su, L.; Ji, S.; Zhang, S.; Yu, P.; Zheng, Y.; Zhang, Q. Inhibition of hepatic stellate cell activation and liver fibrosis by fat-specific protein 27. Mol. Cell. Biochem. 2012, 369, 35–43. [Google Scholar] [CrossRef]
- Haemmerle, G.; Lass, A. Genetically modified mouse models to study hepatic neutral lipid mobilization. Biochim. et Biophys. Acta Mol. Basis Dis. 2019, 1865, 879–894. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e10. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA:cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. BioMed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van Grunsven, L. 3D in vitro models of liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; Van Es, J.H.; Li, V.S.W.; Van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; Van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.C.; Van Wenum, M.; Fuchs, S.A.; De Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]
- Nantasanti, S.; Spee, B.; Kruitwagen, H.S.; Chen, C.; Geijsen, N.; Oosterhoff, L.A.; Van Wolferen, M.E.; Pelaez, N.; Fieten, H.; Wubbolts, R.W.; et al. Disease Modeling and Gene Therapy of Copper Storage Disease in Canine Hepatic Organoids. Stem Cell Rep. 2015, 5, 895–907. [Google Scholar] [CrossRef]
- Kuijk, E.; Rasmussen, S.; Blokzijl, F.; Huch, M.; Gehart, H.; Toonen, P.; Begthel, H.; Clevers, H.; Geurts, A.M.; Cuppen, E. Generation and characterization of rat liver stem cell lines and their engraftment in a rat model of liver failure. Sci. Rep. 2016, 6, 22154. [Google Scholar] [CrossRef]
- Kruitwagen, H.S.; Oosterhoff, L.A.; Vernooij, I.G.; Schrall, I.M.; Van Wolferen, M.E.; Bannink, F.; Roesch, C.; Van Uden, L.; Molenaar, M.; Helms, J.B.; et al. Long-Term Adult Feline Liver Organoid Cultures for Disease Modeling of Hepatic Steatosis. Stem Cell Rep. 2017, 8, 822–830. [Google Scholar] [CrossRef]
- Haaker, M.W.; Kruitwagen, H.S.; Vaandrager, A.B.; Houweling, M.; Penning, L.C.; Molenaar, M.R.; Van Wolferen, M.E.; Oosterhoff, L.A.; Spee, B.; Helms, J.B. Identification of potential drugs for treatment of hepatic lipidosis in cats using an in vitro feline liver organoid system. J. Vet. Intern. Med. 2019, 34, 132–138. [Google Scholar] [CrossRef]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.M.; Noor, F.; Chesne, C.; Van Grunsven, L. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdú, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; E Lavine, J.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Edwards, J.E.; LaCerte, C.; Peyret, T.; Gosselin, N.H.; Marier, J.F.; Hofmann, A.F.; Shapiro, D. Modeling and Experimental Studies of Obeticholic Acid Exposure and the Impact of Cirrhosis Stage. Clin. Transl. Sci. 2016, 9, 328–336. [Google Scholar] [CrossRef]
- Middleton, M.S.; Heba, E.R.; Hooker, C.A.; Bashir, M.R.; Fowler, K.J.; Sandrasegaran, K.; Brunt, E.; Kleiner, D.E.; Doo, E.; Van Natta, M.L.; et al. Agreement Between Magnetic Resonance Imaging Proton Density Fat Fraction Measurements and Pathologist-Assigned Steatosis Grades of Liver Biopsies From Adults With Nonalcoholic Steatohepatitis. Gastroenterology 2017, 153, 753–761. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the New Dual PPARα/δ Agonist GFT505 on Lipid and Glucose Homeostasis in Abdominally Obese Patients with Combined Dyslipidemia or Impaired Glucose Metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noël, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gómez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator−Activated Receptor−α and −δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed]
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol. Metab. 2019, 30, 701–709. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molenaar, M.R.; Penning, L.C.; Helms, J.B. Playing Jekyll and Hyde—The Dual Role of Lipids in Fatty Liver Disease. Cells 2020, 9, 2244. https://doi.org/10.3390/cells9102244
Molenaar MR, Penning LC, Helms JB. Playing Jekyll and Hyde—The Dual Role of Lipids in Fatty Liver Disease. Cells. 2020; 9(10):2244. https://doi.org/10.3390/cells9102244
Chicago/Turabian StyleMolenaar, Martijn R., Louis C. Penning, and J. Bernd Helms. 2020. "Playing Jekyll and Hyde—The Dual Role of Lipids in Fatty Liver Disease" Cells 9, no. 10: 2244. https://doi.org/10.3390/cells9102244
APA StyleMolenaar, M. R., Penning, L. C., & Helms, J. B. (2020). Playing Jekyll and Hyde—The Dual Role of Lipids in Fatty Liver Disease. Cells, 9(10), 2244. https://doi.org/10.3390/cells9102244