A Novel Computational Model for Predicting microRNA–Disease Associations Based on Heterogeneous Graph Convolutional Networks


Abstract
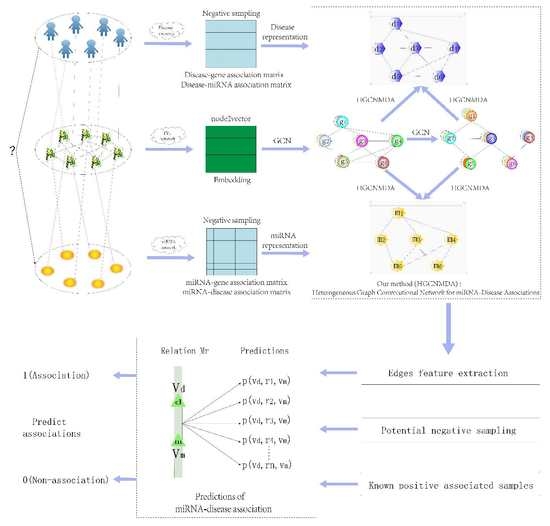
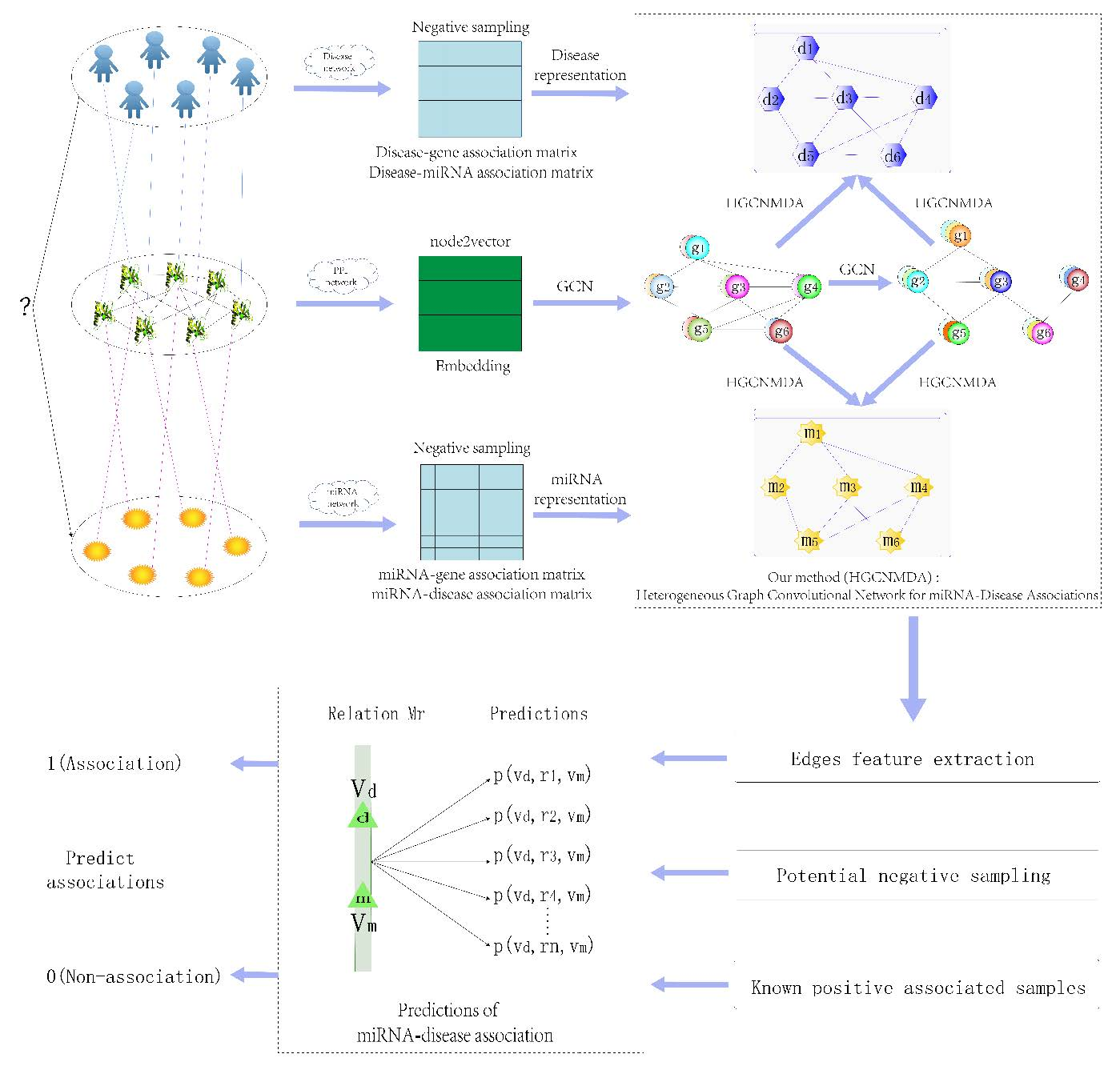
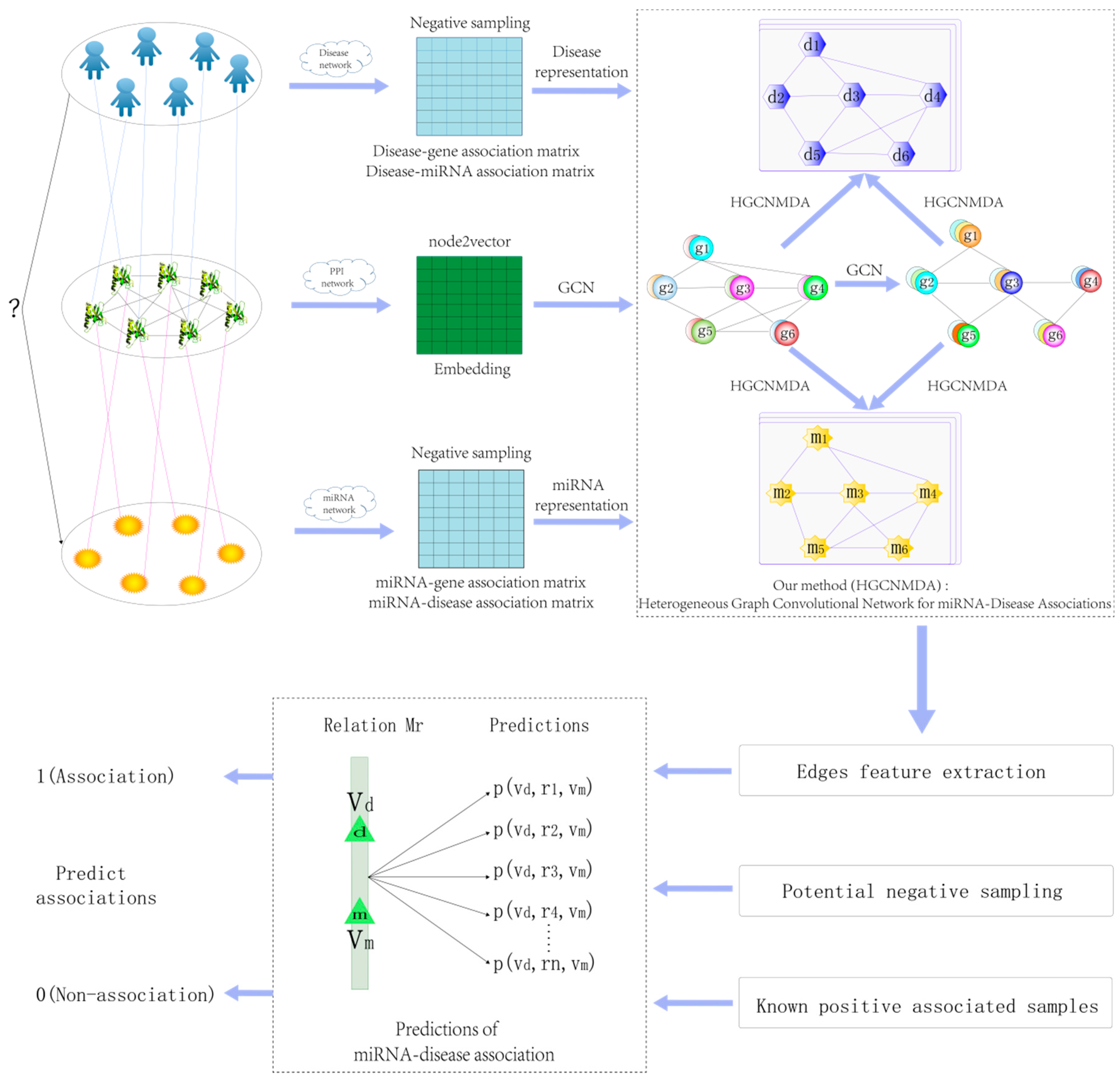
1. Introduction
2. Materials and Methods
2.1. Reconstruction of Heterogeneous Networks
2.1.1. The Human Protein–Protein Interactions
2.1.2. Disease–Gene Network
2.1.3. miRNA–Gene Network
2.1.4. miRNA–Disease Network
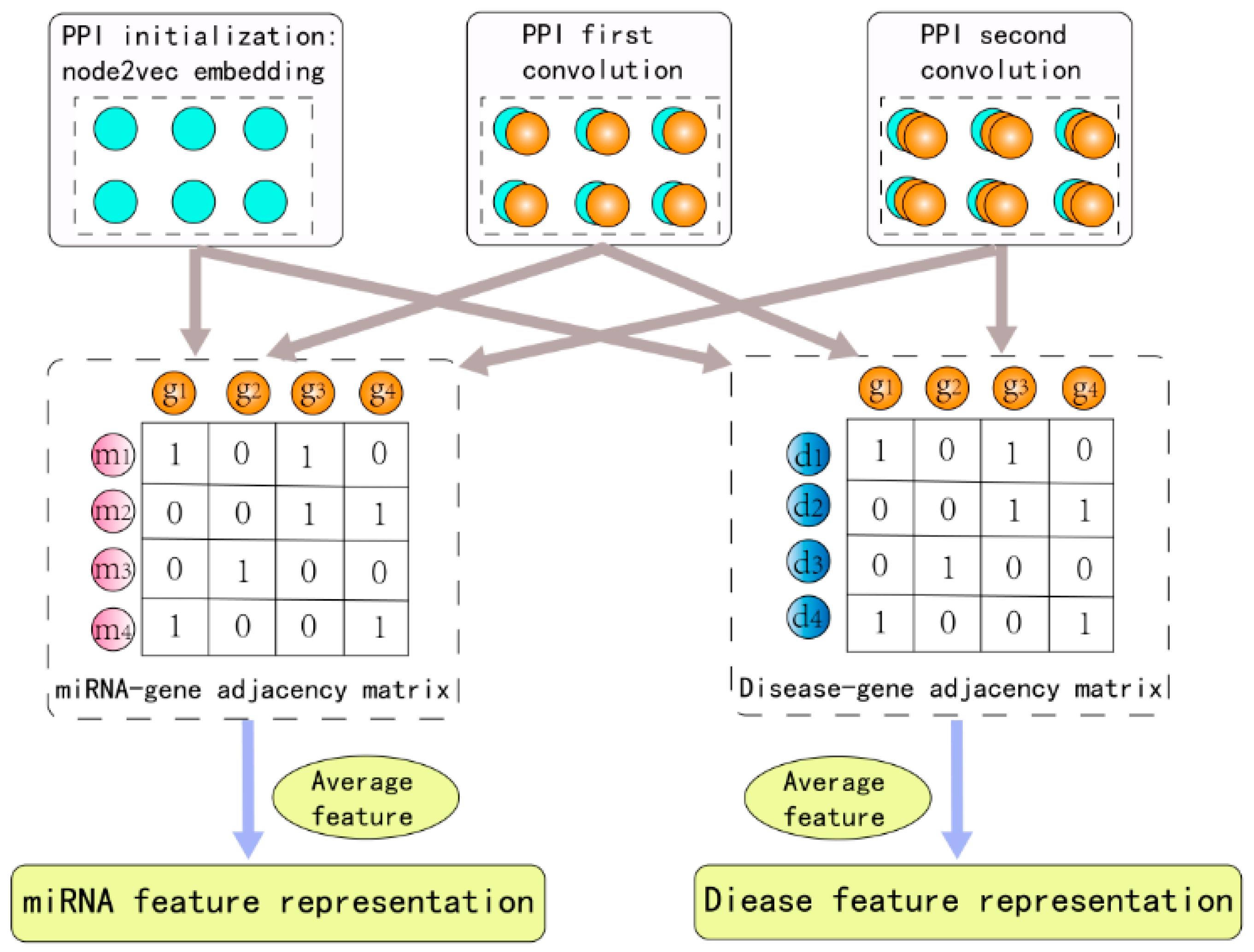
2.2. Raw Feature Extraction
2.3. Graph Convolution Network
2.4. Heterogeneous Graph Convolutional HGCNMDA Approach
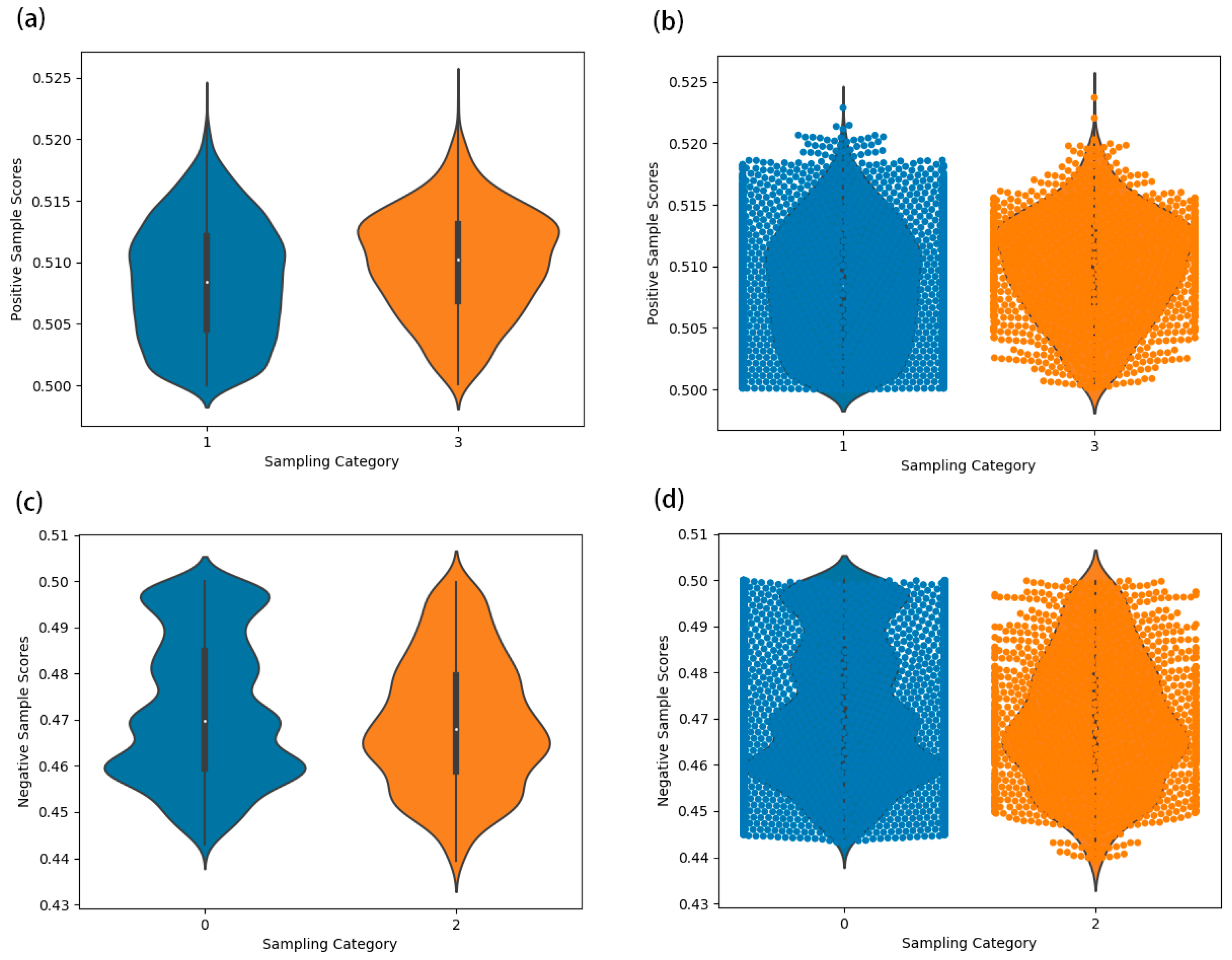
2.4.1. HGCNMDA Convolution Layer and Negative Sampling
2.4.2. Edge Features Extraction
2.4.3. HGCNMDA Model Training
3. Results
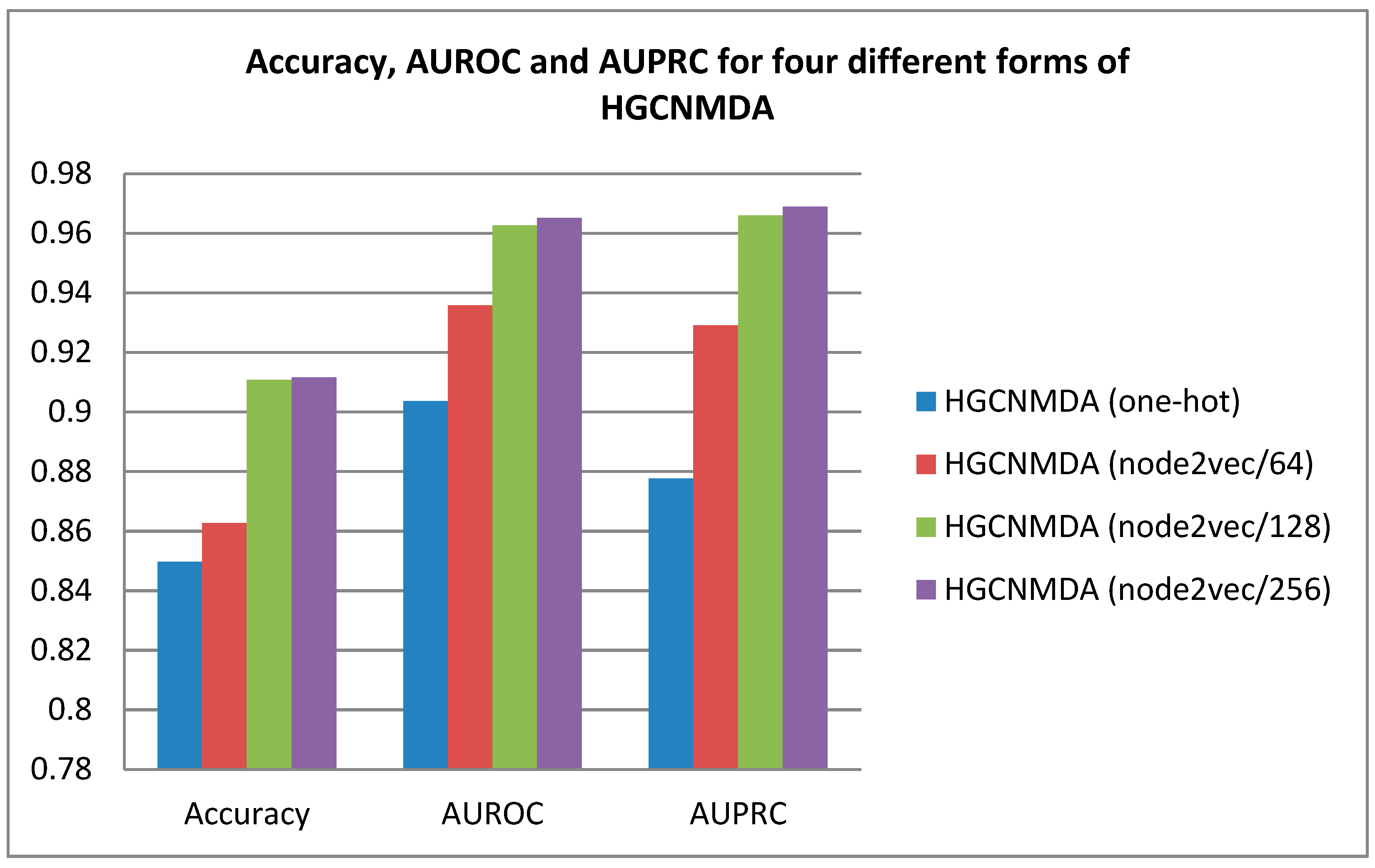
3.1. Overall Performance
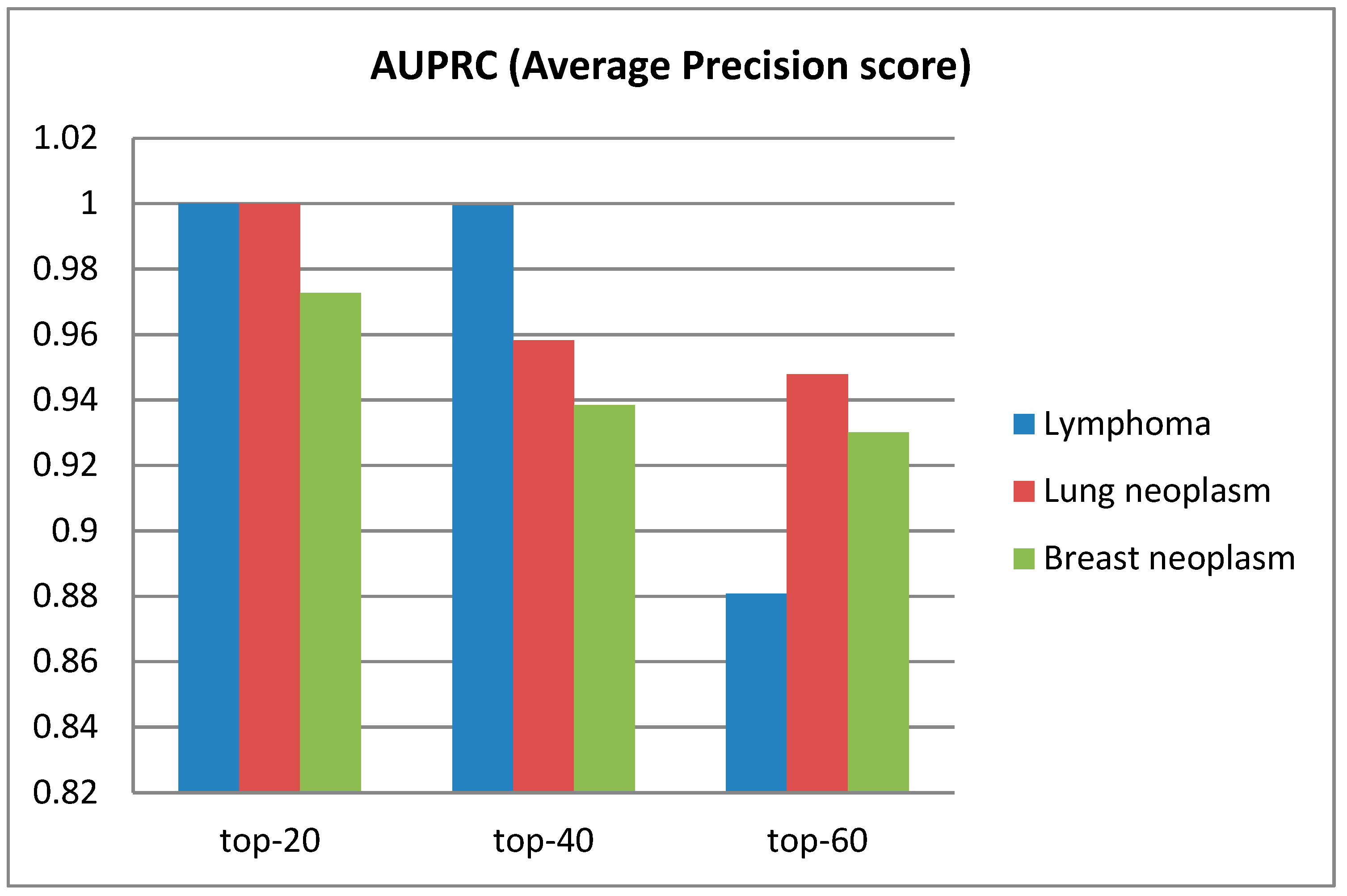
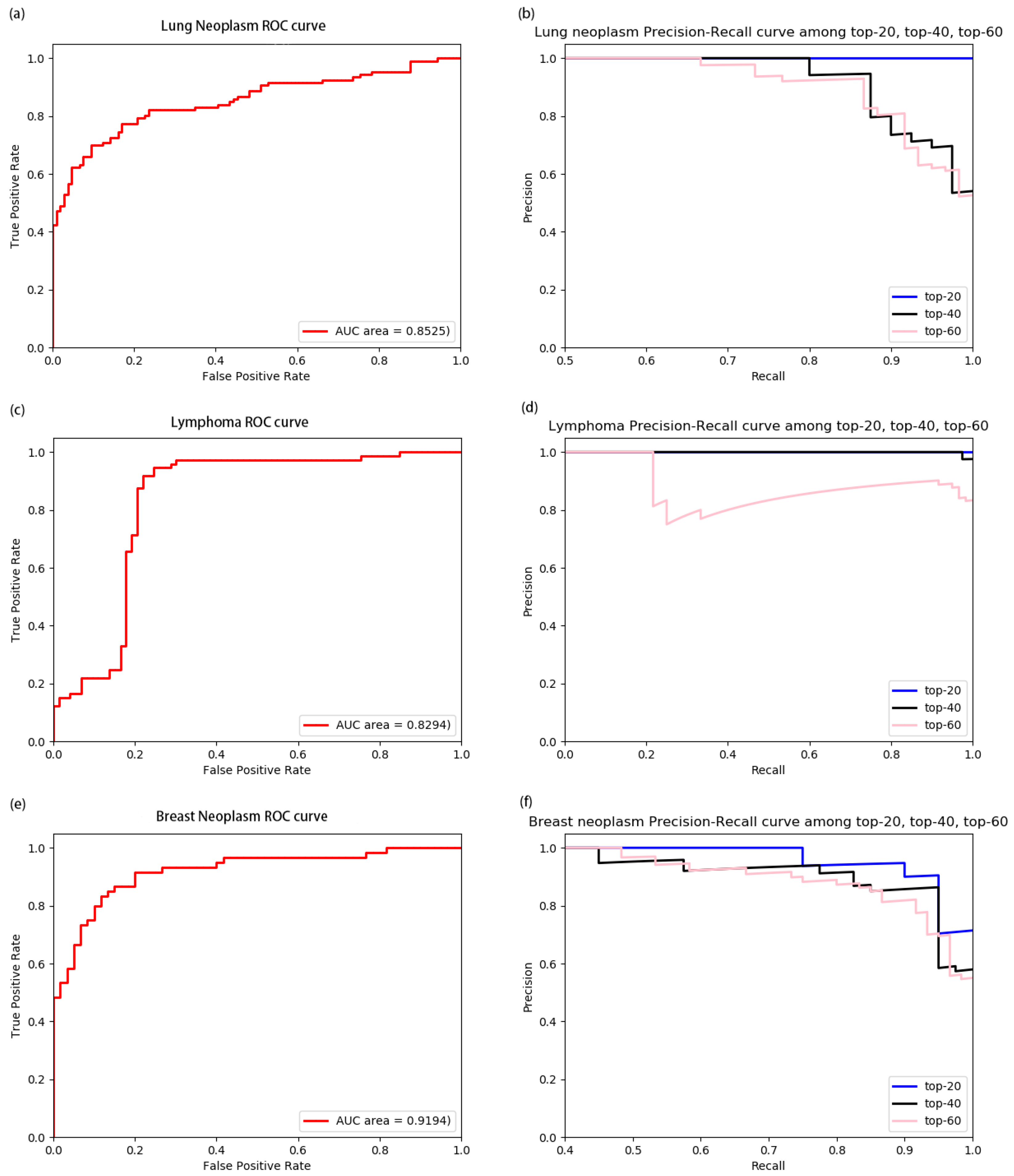
3.2. Performance of Model on Diseases
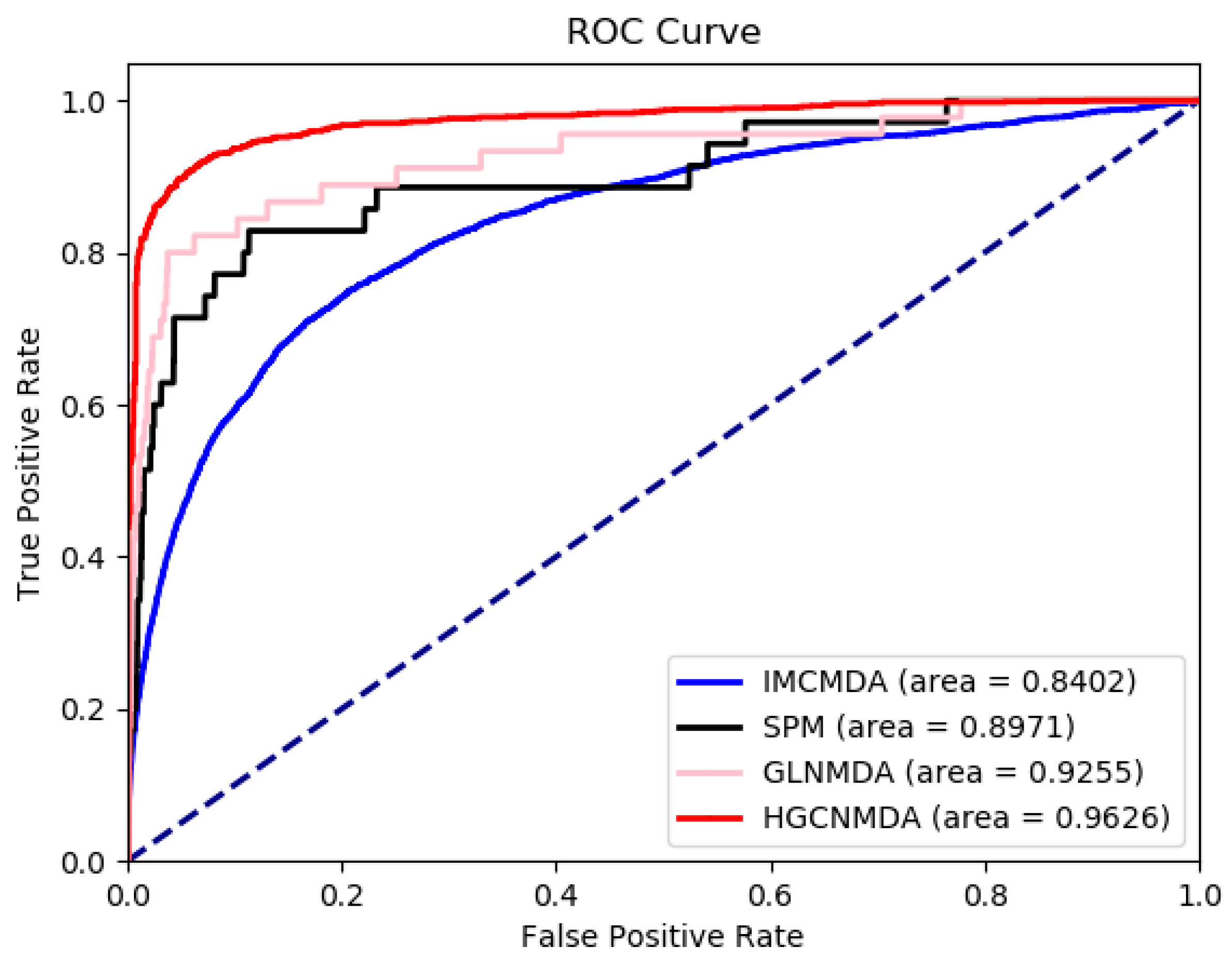
3.3. Comparison To Other Algorithms
3.4. Prediction of New miRNA–Disease Associations
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2015, 17, 193–203. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.-H.; Liu, H. BNPMDA: Bipartite network projection for MiRNA–disease association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.-N.; Li, J.-Q. Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Yu, S.P.; Liang, C.; Xiao, Q.; Li, G.H.; Ding, P.J.; Luo, J.W. GLNMDA: A novel method for miRNA-disease association prediction based on global linear neighborhoods. RNA Biol. 2018, 15, 1215–1227. [Google Scholar] [CrossRef]
- Yu, S.P.; Liang, C.; Xiao, Q.; Li, G.H.; Ding, P.J.; Luo, J.W. MCLPMDA: A novel method for miRNA-disease association prediction based on matrix completion and label propagation. J. Cell. Mol. Med. 2019, 23, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, L.; Lü, L.; Zou, Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Chen, X.; Sun, Y.Z.; Li, J.Q.; Ming, Z. Inferring potential small molecule–miRNA association based on triple layer heterogeneous network. J. Cheminform. 2018, 10, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Q.; Rodriguez Paton, A.; Zeng, X. Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, J.; Song, L.; Zeng, X.; Wang, G. Similarity computation strategies in the microRNA-disease network: A survey. Brief. Funct. Genomics 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Liao, Y.; Pan, L. Prediction and validation of association between microRNAs and diseases by multipath methods. Biochim. Biophys. Acta 2016, 1860, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Li, Y.; Tian, L.P.; Wu, F.X. Enhancing the prediction of disease–gene associations with multimodal deep learning. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Zhang, Z.; Yang, C.; Liu, Z.; Sun, M. Graph neural networks: A review of methods and applications. arXiv 2018, arXiv:08434. [Google Scholar]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:02907. [Google Scholar]
- Grover, A.; Leskovec, J. node2vec: Scalable feature learning for networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: New York, NY, USA, 2016; pp. 855–864. [Google Scholar]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.-L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2014, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33 (Suppl. 1), D514–D517. [Google Scholar] [CrossRef]
- Yu, W.; Gwinn, M.; Clyne, M.; Yesupriya, A.; Khoury, M.J. A navigator for human genome epidemiology. Nat. Genet. 2008, 40, 124–125. [Google Scholar] [CrossRef]
- Hernandez-Boussard, T.; Whirl-Carrillo, M.; Hebert, J.M.; Gong, L.; Owen, R.; Gong, M.; Gor, W.; Liu, F.; Truong, C.; Whaley, R. The pharmacogenetics and pharmacogenomics knowledge base: Accentuating the knowledge. Nucleic Acids Res. 2007, 36 (Suppl. 1), D913–D918. [Google Scholar] [CrossRef]
- Davis, A.P.; King, B.L.; Mockus, S.; Murphy, C.G.; Saraceni-Richards, C.; Rosenstein, M.; Wiegers, T.; Mattingly, C.J. The comparative toxicogenomics database: Update 2011. Nucleic Acids Res. 2010, 39 (Suppl. 1), D1067–D1072. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2013, 41, D8–D20. [Google Scholar] [CrossRef]
- Hsu, S.D.; Tseng, Y.T.; Shrestha, S.; Lin, Y.L.; Khaleel, A.; Chou, C.H.; Chu, C.F.; Huang, H.Y.; Lin, C.M.; Ho, S.Y. miRTarBase update 2014: An information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014, 42, D78–D85. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2008, 37 (Suppl. 1), D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3. 0: A database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2018, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Y. Link prediction based on graph neural networks. In Proceedings of the 32nd International Conference on Neural Information Processing Systems, Montréal, Canada, 3–8 December 2018; pp. 5171–5181. [Google Scholar]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. Deepwalk: Online learning of social representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; ACM: New York, NY, USA; pp. 701–710. [Google Scholar]
- Koren, Y.; Bell, R.; Volinsky, C. Matrix factorization techniques for recommender systems. Computer 2009, 8, 30–37. [Google Scholar] [CrossRef]
- Airoldi, E.M.; Blei, D.M.; Fienberg, S.E.; Xing, E.P. Mixed membership stochastic blockmodels. J. Mach. Learn. Res. 2008, 9, 1981–2014. [Google Scholar] [PubMed]
- Qiu, J.; Dong, Y.; Ma, H.; Li, J.; Wang, K.; Tang, J. Network embedding as matrix factorization: Unifying deepwalk, line, pte, and node2vec. In Proceedings of the 11th ACM International Conference on Web Search and Data Mining, Marina Del Rey, Marina Del Rey, CA, USA, 5–9 February 2018; ACM: New York, NY, USA; pp. 459–467. [Google Scholar]
- Nickel, M.; Jiang, X.; Tresp, V. Reducing the rank in relational factorization models by including observable patterns. In Proceedings of the 27th International Conference on Neural Information Processing Systems, Montreal, Canada, 8–13 December 2014; pp. 1179–1187. [Google Scholar]
- Hammond, D.K.; Vandergheynst, P.; Gribonval, R. Wavelets on graphs via spectral graph theory. Appl. Comput. Harmon. Anal. 2011, 30, 129–150. [Google Scholar] [CrossRef]
- Luo, P.; Tian, L.P.; Ruan, J.; Wu, F.X. Disease gene prediction by integrating ppi networks, clinical rna-seq data and omim data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 222–232. [Google Scholar] [CrossRef]
- Yang, P.; Li, X.L.; Mei, J.P.; Kwoh, C.K.; Ng, S.K. Positive-unlabeled learning for disease gene identification. Bioinformatics 2012, 28, 2640–2647. [Google Scholar] [CrossRef]
- Nickel, M.; Tresp, V.; Kriegel, H.-P. A Three-Way Model for Collective Learning on Multi-Relational Data. In Proceedings of the 28th International Conference on International Conference on Machine Learning, Bellevue, WA, USA, 28 June–2 July 2011; pp. 809–816. [Google Scholar]
- Trouillon, T.; Welbl, J.; Riedel, S.; Gaussier, É.; Bouchard, G. Complex embeddings for simple link prediction. In Proceedings of the International Conference on Machine Learning, New York, NY, USA, 19–24 June 2016; pp. 2071–2080. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Glorot, X.; Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. In Proceedings of the 13th International Conference on Artificial Intelligence and Statistics, Sardinia, Italy, 13–15 May 2010; pp. 249–256. [Google Scholar]
- Li, Q.; Han, Z.; Wu, X.M. Deeper insights into graph convolutional networks for semi-supervised learning. In Proceedings of the 32nd AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018; pp. 3538–3545. [Google Scholar]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, A.; Wissing, M.; Salö, S.; Englund, A.; Dalgaard, L. MicroRNAs related to polycystic ovary syndrome (PCOS). Genes 2014, 5, 684–708. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.Y.; Wu, H.L.; Chen, C.C.; Gamboa, G.M.; Layman, L.C.; Diamond, M.P.; Azziz, R.; Chen, Y.H. MicroRNA-223 expression is upregulated in insulin resistant human adipose tissue. J. Diabet. Res. 2015, 2015, 943659. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Ma, X.; Chen, B.; Huang, Y.; Liu, S.; Yang, H.; Zou, W. MicroRNA-145 negatively regulates cell proliferation through targeting IRS1 in isolated ovarian granulosa cells from patients with polycystic ovary syndrome. Reprod. Sci. 2017, 24, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.W.; McCallie, B.; Alvero, R.; Schoolcraft, W.B.; Minjarez, D.; Katz-Jaffe, M.G. Altered microRNA and gene expression in the follicular fluid of women with polycystic ovary syndrome. J. Assist. Reprod. Genet. 2014, 31, 355–362. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Baselines | Accuracy | AUROC | AUPRC |
|---|---|---|---|
| HGCNMDA (One-hot) | 0.8497 | 0.9036 | 0.8776 |
| HGCNMDA (Node2vec/64) | 0.8626 | 0.9358 | 0.9290 |
| HGCNMDA (Node2vec/128) | 0.9108 | 0.9626 | 0.9660 |
| HGCNMDA (Node2vec/256) | 0.9116 | 0.9651 | 0.9689 |
| Osteosarcoma | Polycystic Ovary Syndrome | ||
|---|---|---|---|
| miRNA | Evidence | miRNA | Evidence |
| hsa-mir-26b | dbDEMCv2.0 | hsa-mir-9 | (Sørensen et al., 2014) [50] |
| hsa-mir-218 | Unconfirmed | hsa-mir-21 | (Sørensen et al., 2014) [50] |
| hsa-mir-873 | Unconfirmed | hsa-mir-155 | (Sørensen et al., 2014) [50] |
| hsa-mir-383 | dbDEMCv2.0 | hsa-mir-146a | (Sørensen et al., 2014) [50] |
| hsa-mir-16 | dbDEMCv2.0 | hsa-mir-223 | (Chuang et al., 2015) [51] |
| hsa-mir-199a | dbDEMCv2.0 | hsa-mir-34a | Unconfirmed |
| hsa-mir-671 | dbDEMCv2.0 | hsa-mir-145 | (Cai et al., 2017) [52] |
| hsa-mir-367 | dbDEMCv2.0 | hsa-mir-126 | Unconfirmed |
| hsa-mir-145 | dbDEMCv2.0 | hsa-mir-210 | Unconfirmed |
| hsa-mir-17 | dbDEMCv2.0 | hsa-mir-32 | (Roth et al., 2014) [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Liu, H.; Hu, Q.; Que, J.; Yao, J. A Novel Computational Model for Predicting microRNA–Disease Associations Based on Heterogeneous Graph Convolutional Networks. Cells 2019, 8, 977. https://doi.org/10.3390/cells8090977
Li C, Liu H, Hu Q, Que J, Yao J. A Novel Computational Model for Predicting microRNA–Disease Associations Based on Heterogeneous Graph Convolutional Networks. Cells. 2019; 8(9):977. https://doi.org/10.3390/cells8090977
Chicago/Turabian StyleLi, Chunyan, Hongju Liu, Qian Hu, Jinlong Que, and Junfeng Yao. 2019. "A Novel Computational Model for Predicting microRNA–Disease Associations Based on Heterogeneous Graph Convolutional Networks" Cells 8, no. 9: 977. https://doi.org/10.3390/cells8090977
APA StyleLi, C., Liu, H., Hu, Q., Que, J., & Yao, J. (2019). A Novel Computational Model for Predicting microRNA–Disease Associations Based on Heterogeneous Graph Convolutional Networks. Cells, 8(9), 977. https://doi.org/10.3390/cells8090977