Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Microscopy
2.2. C. elegans Strains, Maintenance, and Methods
2.3. Quantitative Real-Time PCR
2.4. Statistical Methods
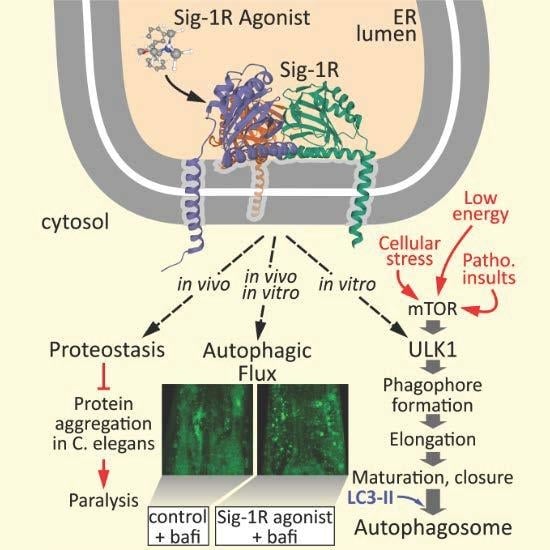
3. Results and Discussion
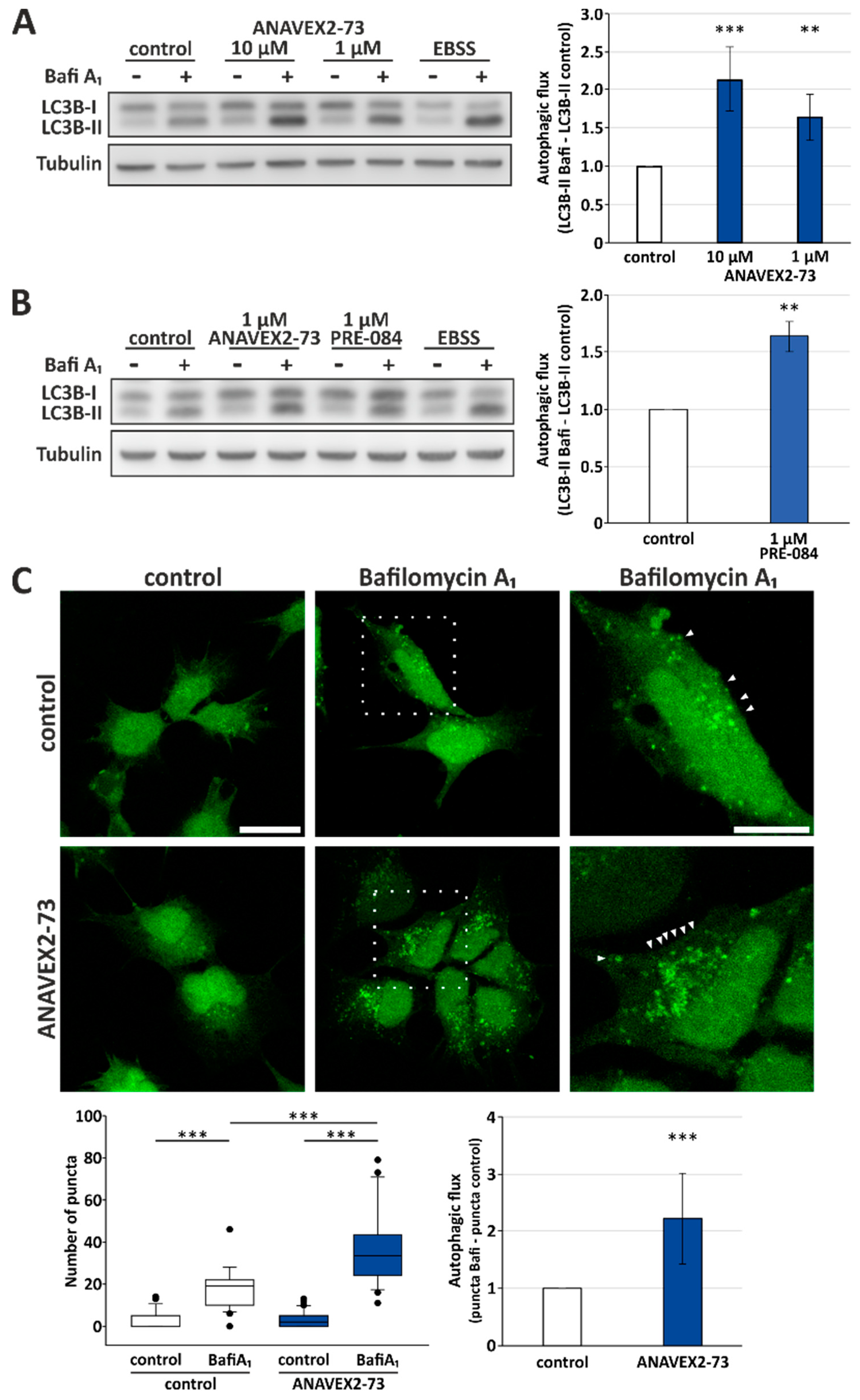
3.1. Sig1-R Agonist ANAVEX2-73 Enhances Autophagic Activity
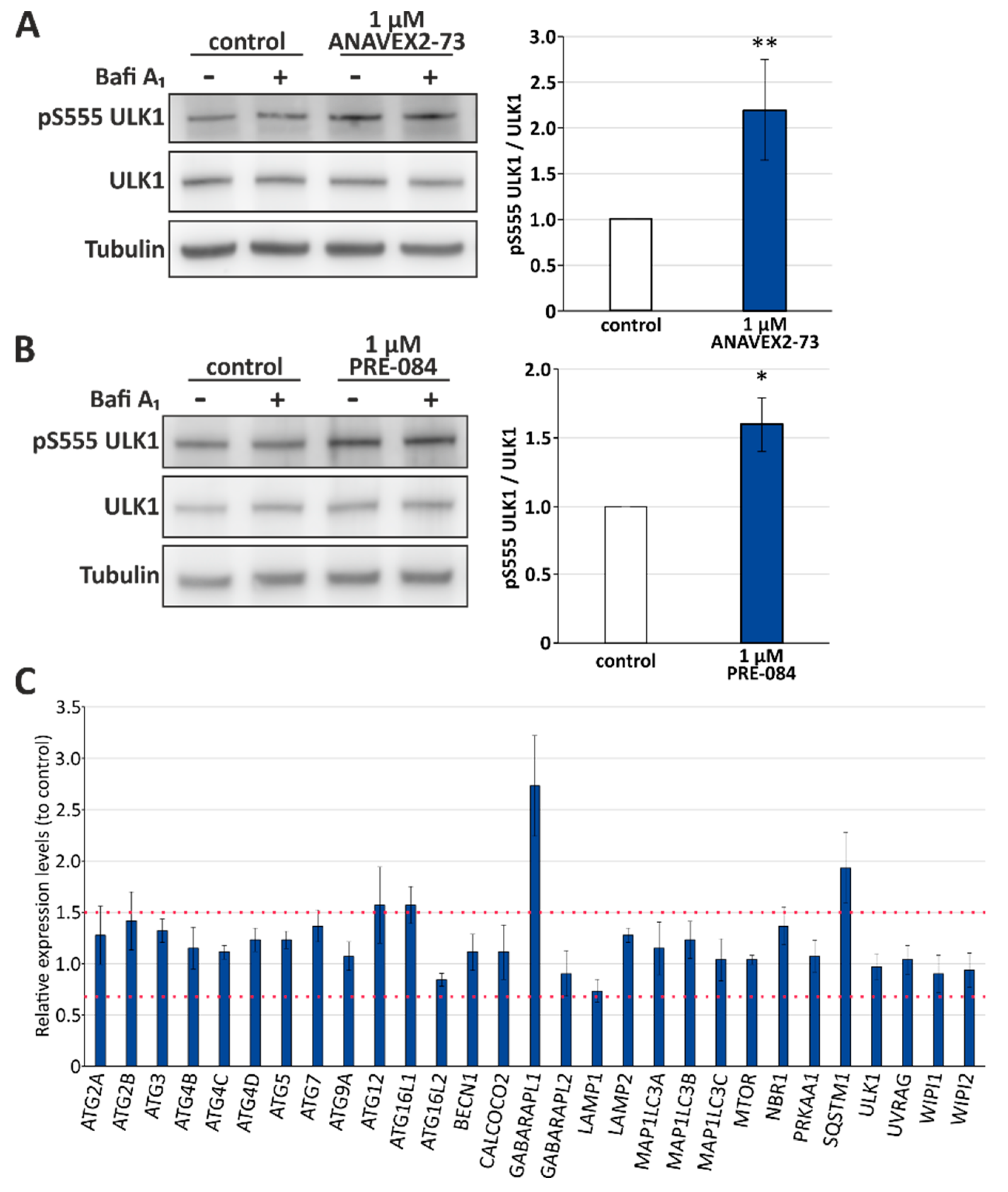
3.2. Sig-1R Activation Induces ULK1 Phosphorylation and Affects Expression Levels of Distinct Autophagy Network Factors
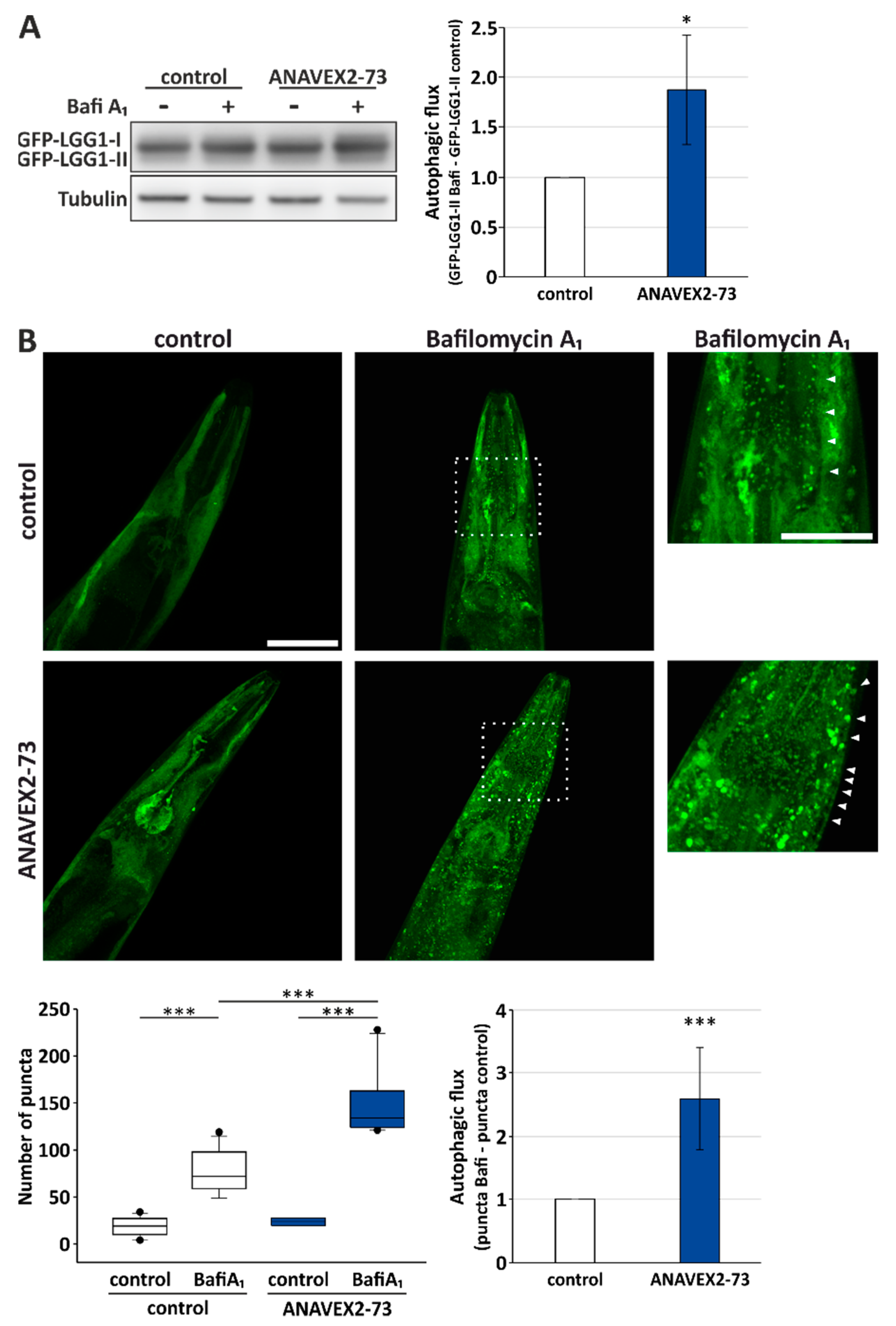
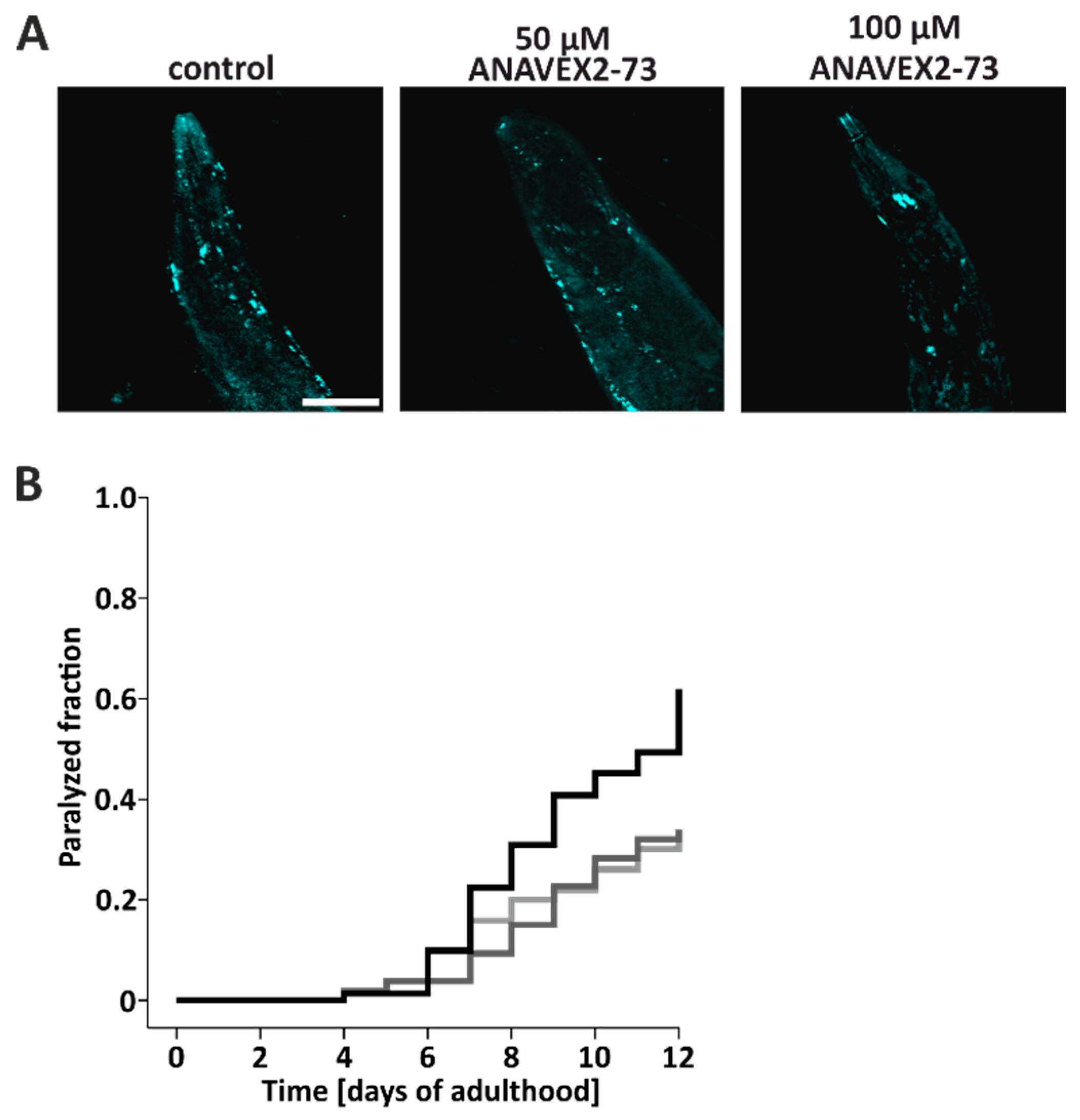
3.3. ANAVEX2-72 Positively Regulates Autophagy, Increases Proteostasis Capacity, and Improves Protein Aggregation-Mediated Paralysis in C. elegans
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein homeostasis, aging and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, H.; Klionsky, D.J. Autophagy in yeast: Mechanistic insights and physiological function. Microbiol. Mol. Biol. Rev. 2001, 65, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. Adv. Exp. Med. Biol. 2017, 964, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.W.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [PubMed]
- Kekuda, R.; Prasad, P.D.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V. Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem. Biophys. Res. Commun. 1996, 229, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.A.; Su, T.P. Sigma-1 Receptors Fine-Tune the Neuronal Networks. Adv. Exp. Med. Biol. 2017, 964, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Brune, S.; Schepmann, D.; Klempnauer, K.H.; Marson, D.; Dal Col, V.; Laurini, E.; Fermeglia, M.; Wunsch, B.; Pricl, S. The sigma enigma: In vitro/in silico site-directed mutagenesis studies unveil sigma1 receptor ligand binding. Biochemistry 2014, 53, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Goguadze, N. Role of sigma1 Receptors in Learning and Memory and Alzheimer’s Disease-Type Dementia. Adv. Exp. Med. Biol. 2017, 964, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Vamvakides, A. [Anticonvulsant and forced swim anti-immobility effects of tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanemethanamine (AE37): Common action mechanism?]. Ann. Pharm. Fr. 2002, 60, 88–92. [Google Scholar] [PubMed]
- Espallergues, J.; Lapalud, P.; Christopoulos, A.; Avlani, V.A.; Sexton, P.M.; Vamvakides, A.; Maurice, T. Involvement of the sigma1 (sigma1) receptor in the anti-amnesic, but not antidepressant-like, effects of the aminotetrahydrofuran derivative ANAVEX1-41. Br. J. Pharmacol. 2007, 152, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Parmentier, F.; Ette, E.I.; Fadiran, E.O.; Missling, C.U. Clinical Pharmacokinetics and Pharmacodynamics Characterization of ANAVEX™2-73 for Designing a Phase 2/3 Study in Mild-to-Moderate Alzheimer’s Disease. In Proceedings of the 10th Clinical Trials on Alzheimer’s Disease, Boston, MA, USA, 4 November 2017. [Google Scholar]
- Hampel, H.; Afshar, M.; Parmentier, F.; Williams, C.; Etcheto, A.; Goodsaid, F.; Missling, C.U. Longitudinal 148-Week Extension Study for ANAVEX®2-73 Phase 2a Alzheimer’s Disease Demonstrates Maintained Activities of Daily Living Score (ADCS-ADL) and Reduced Cognitive Decline (MMSE) for Patient Cohort on Higher Drug Concentration and Confirms Role of Patient Selection Biomarkers. In Proceedings of the 11th Clinical Trials on Alzheimer’s Disease, Barcellona, Spain, 24–27 October 2018. [Google Scholar]
- Hampel, H.; Afshar, M.; Parmentier, F.; Williams, C.; Etcheto, A.; Goodsaid, F.; Fadiran, E.O.; Missling, C.U. Full Genomic Analysis of ANAVEX®2-73 Phase 2a Alzheimer’s Disease Study Identifies Biomarkers Enabling Targeted Therapy and a Precision Medicine Approach. In Proceedings of the Alzheimer’s Association International Conference 2018, Chicago, IL, USA, 22–26 July 2018. [Google Scholar]
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/sigma1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Abeta25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2014, 8, 463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Mysona, B.A.; Wang, J.; Gonsalvez, G.B.; Smith, S.B.; Bollinger, K.E. Sigma 1 receptor regulates ERK activation and promotes survival of optic nerve head astrocytes. PLoS ONE 2017, 12, e0184421. [Google Scholar] [CrossRef] [PubMed]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 2014, 5, e1290. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Walker, M.P.; Vaidya, N.K.; Fu, M.; Kumar, S.; Kumar, A. Cocaine-Mediated Autophagy in Astrocytes Involves Sigma 1 Receptor, PI3K, mTOR, Atg5/7, Beclin-1 and Induces Type II Programed Cell Death. Mol. Neurobiol. 2016, 53, 4417–4430. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Gamerdinger, M.; Hajieva, P.; Kaya, A.M.; Wolfrum, U.; Hartl, F.U.; Behl, C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009, 28, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Spang, N.; Feldmann, A.; Huesmann, H.; Bekbulat, F.; Schmitt, V.; Hiebel, C.; Koziollek-Drechsler, I.; Clement, A.M.; Moosmann, B.; Jung, J.; et al. RAB3GAP1 and RAB3GAP2 modulate basal and rapamycin-induced autophagy. Autophagy 2014, 10, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Bekbulat, F.; Huesmann, H.; Ulbrich, S.; Tatzelt, J.; Behl, C.; Kern, A. The RAB GTPase RAB18 modulates macroautophagy and proteostasis. Biochem. Biophys. Res. Commun. 2017, 486, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Spang, N.; Huesmann, H.; Behl, C. Novel Modulators of Proteostasis: RNAi Screen of Chromosome I in a Heat Stress Paradigm in C. elegans. Cells 2018, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Entrena, J.M.; Sanchez-Fernandez, C.; Nieto, F.R.; Gonzalez-Cano, R.; Yeste, S.; Cobos, E.J.; Baeyens, J.M. Sigma-1 Receptor Agonism Promotes Mechanical Allodynia After Priming the Nociceptive System with Capsaicin. Sci. Rep. 2016, 6, 37835. [Google Scholar] [CrossRef] [PubMed]
- Joachim, J.; Jiang, M.; McKnight, N.C.; Howell, M.; Tooze, S.A. High-throughput screening approaches to identify regulators of mammalian autophagy. Methods 2015, 75, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, L.; Yu, X.J.; Miao, Y.; Liu, J.J.; Wang, H.; Ren, J.; Zang, W.J. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 32, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Klionsky, D.J. AMPK activates autophagy by phosphorylating ULK1. Circ. Res. 2011, 108, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Chakrama, F.Z.; Seguin-Py, S.; Le Grand, J.N.; Fraichard, A.; Delage-Mourroux, R.; Despouy, G.; Perez, V.; Jouvenot, M.; Boyer-Guittaut, M. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010, 6, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Epstein, M.L.; Verbny, Y.I.; Huerta, M.S.; Zaitoun, I.; Ziskind-Conhaim, L.; Ruoho, A.E. Lack of sigma-1 receptor exacerbates ALS progression in mice. Neuroscience 2013, 240, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Olivan, S.; Rando, A.; Casas, C.; Osta, R.; Navarro, X. Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 2012, 9, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Gregianin, E.; Pallafacchina, G.; Zanin, S.; Crippa, V.; Rusmini, P.; Poletti, A.; Fang, M.; Li, Z.; Diano, L.; Petrucci, A.; et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 2016, 25, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Reviews. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.L.; Fang, M.; Zhao, Y.X.; Liu, X.Y. Roles of sigma-1 receptors in Alzheimer’s disease. Int. J. Clin. Exp. Med. 2015, 8, 4808–4820. [Google Scholar] [PubMed]
- Penke, B.; Fulop, L.; Szucs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (sigma1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2011, 25, 1101–1117. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christ, M.G.; Huesmann, H.; Nagel, H.; Kern, A.; Behl, C. Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo. Cells 2019, 8, 211. https://doi.org/10.3390/cells8030211
Christ MG, Huesmann H, Nagel H, Kern A, Behl C. Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo. Cells. 2019; 8(3):211. https://doi.org/10.3390/cells8030211
Chicago/Turabian StyleChrist, Maximilian G., Heike Huesmann, Heike Nagel, Andreas Kern, and Christian Behl. 2019. "Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo" Cells 8, no. 3: 211. https://doi.org/10.3390/cells8030211
APA StyleChrist, M. G., Huesmann, H., Nagel, H., Kern, A., & Behl, C. (2019). Sigma-1 Receptor Activation Induces Autophagy and Increases Proteostasis Capacity In Vitro and In Vivo. Cells, 8(3), 211. https://doi.org/10.3390/cells8030211