Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications



Abstract
1. Introduction
2. Determinants of Metabolic Reprogramming
2.1. Cell Intrinsic Cues: Role of Estrogen Receptors and p53 in Breast Cancer
2.1.1. Role of ERα in Regulating Breast Cancer Metabolism
2.1.2. Role of ERβ in Regulating Breast Cancer Metabolism
2.1.3. Role of p53 in Regulating Cancer Metabolism
2.1.4. ER-p53 Crosstalk and its Implications in Breast Cancer Metabolism
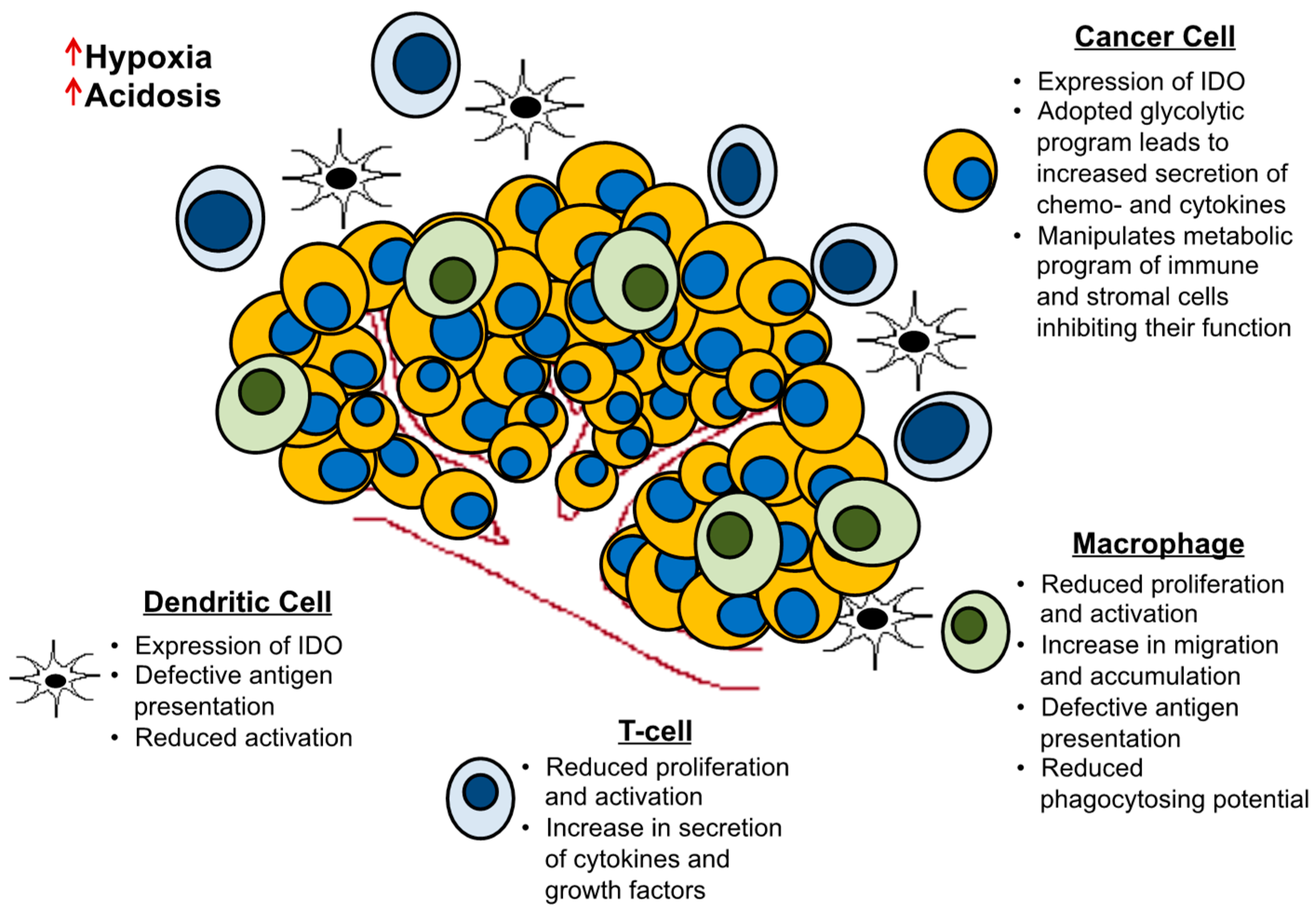
2.2. Cell Extrinsic Cues and Metabolic Interactions with the Tumor Microenvironment
2.3. Cytoplasmic/Mitochondrial-Nuclear Crosstalk Regulates Breast Cancer Metabolism
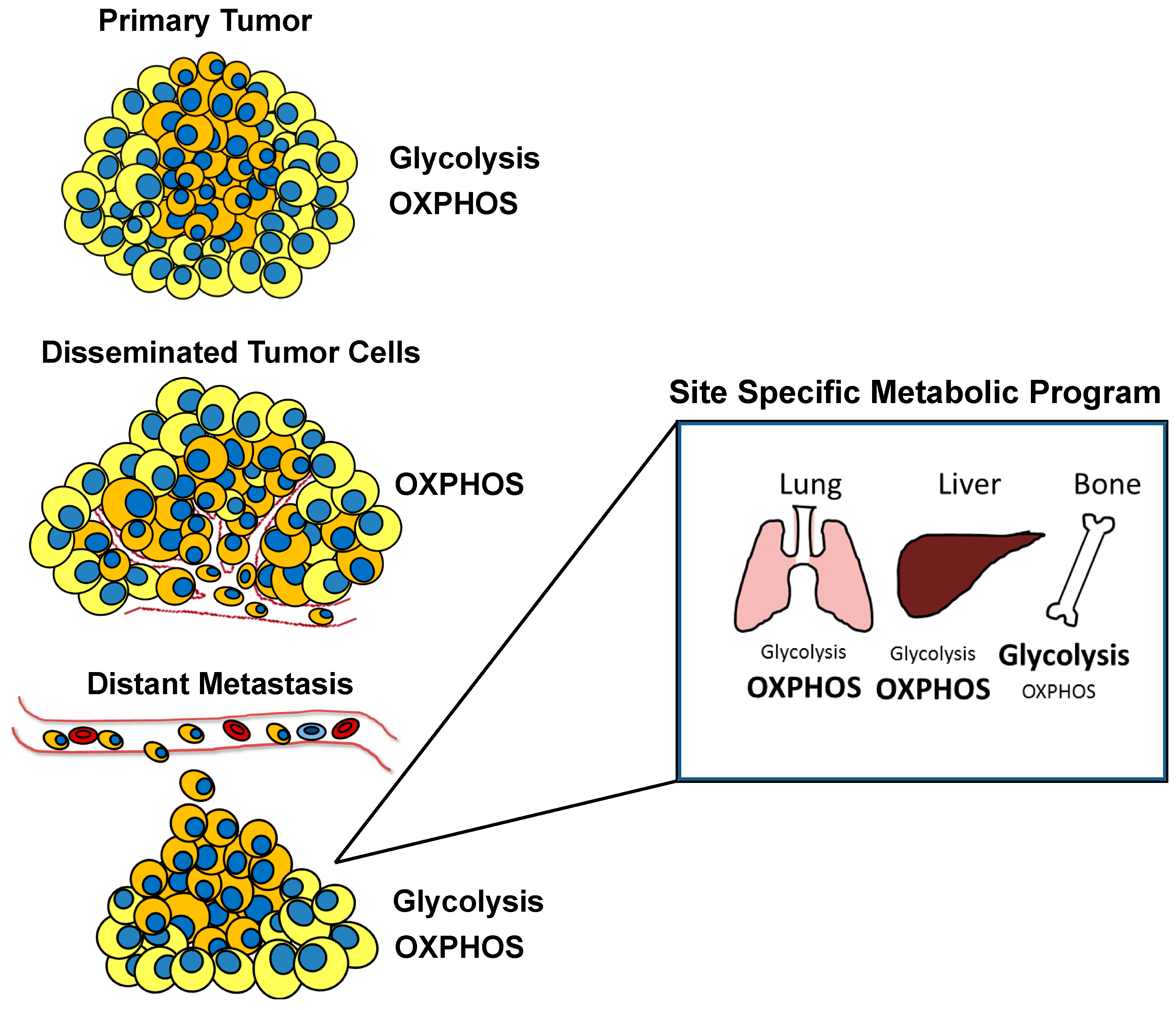
3. Altered Metabolism in Primary versus Metastatic Lesions and Site-Specific Metabolic Alterations
4. Effects of Treatment and Resistance to SOC on Metabolic Reprogramming
4.1. Luminal A/B Subtype
4.2. HER2-Enriched Breast Cancer
4.3. TNBC/Basal-Like-Cancer
5. Current Status of Metabolic Intervention in Breast Cancer: Challenges that Lie Ahead
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Rev. Cancer 2016, 16, 613–614. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Dupuy, F.; Bastien Tabariè, S.; Andrzejewski, S.; Pierre, J.S.; Jones, R.G.; Siegel, P.M.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Simoes, R.V.; Serganova, I.S.; Kruchevsky, N.; Leftin, A.; Shestov, A.A.; Thaler, H.T.; Sukenick, G.; Locasale, J.W.; Blasberg, R.G.; Koutcher, J.A.; et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., III; Felding-Habermann, B. Adaptation of Energy Metabolism in Breast Cancer Brain Metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabaries, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chenard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab 2017, 26, 778–787. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen Receptor Status by Immunohistochemistry Is Superior to the Ligand-Binding Assay for Predicting Response to Adjuvant Endocrine Therapy in Breast Cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef]
- Mohsin, S.K.; Weiss, H.; Havighurst, T.; Clark, G.M.; Berardo, M.; Roanh, L.D.; To, T.V.; Zho, Q.; Love, R.R.; Allred, D.C. Progesterone receptor by immunohistochemistry and clinical outcome in breast cancer: A validation study. Mod. Pathol. 2004, 17, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 2007, 25, 118–145. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Romond, E.H.; Perez, E.A.; Bryant, J.; Suman, V.J.; Geyer, C.E., Jr.; Davidson, N.E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman, P.A.; et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Nahta, R.; Yu, D.; Hung, M.C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [CrossRef]
- Camarda, R.; Williams, J.; Goga, A. In vivo Reprogramming of Cancer Metabolism by MYC. Front. Cell Dev. Biol. 2017, 5, 35. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Olopade, O.I. MYC and Breast Cancer. Genes Cancer 2010, 1, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Wickramasekera, N.T.; Das, G.M. Tumor suppressor p53 and estrogen receptors in nuclear-mitochondrial communication. Mitochondrion 2014, 16, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Cuyas, E.; Fernandez-Arroyo, S.; Alarcon, T.; Lupu, R.; Joven, J.; Menendez, J.A. Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: Evidence for metformin-based "starvation" strategies in BRCA1 carriers. Oncotarget 2016, 7, 52974–52992. [Google Scholar] [CrossRef] [PubMed]
- Koobotse, M.; Holly, J.; Perks, C. Elucidating the novel BRCA1 function as a non-genomic metabolic restraint in ER-positive breast cancer cell lines. Oncotarget 2018, 9, 33562–33576. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.; Lin, Z.; Whitaker-Menezes, D.; Birbe, R.C.; Bombonati, A.; Pavlides, S.; Lamb, R.; Sneddon, S.; Howell, A.; et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: Implications for breast cancer prevention with antioxidant therapies. Cell Cycle 2012, 11, 4402–4413. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Dizin, E.; Ray, H.; Luquain, C.; Lefai, E.; Foufelle, F.; Billaud, M.; Lenoir, G.M.; Venezia, N.D. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J. Biol. Chem. 2006, 281, 3172–3181. [Google Scholar] [CrossRef]
- Privat, M.; Radosevic-Robin, N.; Aubel, C.; Cayre, A.; Penault-Llorca, F.; Marceau, G.; Sapin, V.; Bignon, Y.J.; Morvan, D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS ONE 2014, 9, e102438. [Google Scholar] [CrossRef]
- Vazquez-Arreguin, K.; Maddox, J.; Kang, J.; Park, D.; Cano, R.R.; Factor, R.E.; Ludwig, T.; Tantin, D. BRCA1 through Its E3 Ligase Activity Regulates the Transcription Factor Oct1 and Carbohydrate Metabolism. Mol. Cancer Res. 2018, 16, 439–452. [Google Scholar] [CrossRef]
- Holstege, H.; Horlings, H.M.; Velds, A.; Langerod, A.; Borresen-Dale, A.L.; van de Vijver, M.J.; Nederlof, P.M.; Jonkers, J. BRCA1-mutated and basal-like breast cancers have similar aCGH profiles and a high incidence of protein truncating TP53 mutations. BMC Cancer 2010, 10, 654. [Google Scholar] [CrossRef]
- Kumar, P.; Mukherjee, M.; Johnson, J.P.; Patel, M.; Huey, B.; Albertson, D.G.; Simin, K. Cooperativity of Rb, Brca1, and p53 in malignant breast cancer evolution. PLoS Genet. 2012, 8, e1003027. [Google Scholar] [CrossRef]
- Wang, L.; Di, L.J. BRCA1 and estrogen/estrogen receptor in breast cancer: Where they interact? Int. J. Biol. Sci. 2014, 10, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors alpha and beta in cancer. Crit. Rev. Oncol. Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Skliris, G.P.; Burdall, S.E.; Carder, P.J. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J. Clin. Pathol. 2002, 55, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Forster, C.; Makela, S.; Warri, A.; Kietz, S.; Becker, D.; Hultenby, K.; Warner, M.; Gustafsson, J.A. Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 15578–15583. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Purdie, C.A.; Harrison, D.J.; Peter, A.; Dobbie, L.; White, S.; Howie, S.E.; Salter, D.M.; Bird, C.C.; Wyllie, A.H.; Hooper, M.L.; et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 1994, 9, 603–609. [Google Scholar]
- Li, M.; Hu, J.; Heermeier, K.; Hennighausen, L.; Furth, P.A. Apoptosis and remodeling of mammary gland tissue during involution proceeds through p53-independent pathways. Cell Growth Differ. 1996, 7, 13–20. [Google Scholar]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef]
- Chiche, A.; Moumen, M.; Petit, V.; Jonkers, J.; Medina, D.; Deugnier, M.A.; Faraldo, M.M.; Glukhova, M.A. Somatic loss of p53 leads to stem/progenitor cell amplification in both mammary epithelial compartments, basal and luminal. Stem Cells 2013, 31, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Jerry, D.J.; Kuperwasser, C.; Downing, S.R.; Pinkas, J.; He, C.; Dickinson, E.; Marconi, S.; Naber, S.P. Delayed involution of the mammary epithelium in BALB/c-p53null mice. Oncogene 1998, 17, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Gatza, C.E.; Dumble, M.; Kittrell, F.; Edwards, D.G.; Dearth, R.K.; Lee, A.V.; Xu, J.; Medina, D.; Donehower, L.A. Altered mammary gland development in the p53+/m mouse, a model of accelerated aging. Dev. Biol. 2008, 313, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Kuperwasser, C.; Hurlbut, G.D.; Kittrell, F.S.; Dickinson, E.S.; Laucirica, R.; Medina, D.; Naber, S.P.; Jerry, D.J. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am. J. Pathol. 2000, 157, 2151–2159. [Google Scholar] [CrossRef]
- Medina, D.; Kittrell, F.S. p53 function is required for hormone-mediated protection of mouse mammary tumorigenesis. Cancer Res. 2003, 63, 6140–6143. [Google Scholar] [PubMed]
- Li, B.; Rosen, J.M.; McMenamin-Balano, J.; Muller, W.J.; Perkins, A.S. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Mol. Cell. Biol. 1997, 17, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, D.P.; Xu, Y. The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene 2013, 32, 2900–2906. [Google Scholar] [CrossRef] [PubMed]
- MacMahon, B.; Cole, P.; Lin, T.M.; Lowe, C.R.; Mirra, A.P.; Ravnihar, B.; Salber, E.J.; Valaoras, V.G.; Yuasa, S. Age at first birth and breast cancer risk. Bull. World Health Organ. 1970, 43, 209–221. [Google Scholar] [PubMed]
- Kelsey, J.L.; Gammon, M.D. The epidemiology of breast cancer. CA Cancer J. Clin. 1991, 41, 146–165. [Google Scholar] [CrossRef]
- Lambe, M.; Hsieh, C.; Trichopoulos, D.; Ekbom, A.; Pavia, M.; Adami, H.O. Transient increase in the risk of breast cancer after giving birth. N. Engl. J. Med. 1994, 331, 5–9. [Google Scholar] [CrossRef]
- Albrektsen, G.; Heuch, I.; Hansen, S.; Kvale, G. Breast cancer risk by age at birth, time since birth and time intervals between births: Exploring interaction effects. Br. J. Cancer 2005, 92, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Moon, R.C.; Morii, S. Extinction of experimental mammary cancer. I. Estradiol-17beta and progesterone. Proc. Natl. Acad. Sci. USA 1962, 48, 379–386. [Google Scholar] [CrossRef]
- Grubbs, C.J.; Juliana, M.M.; Whitaker, L.M. Short-term hormone treatment as a chemopreventive method against mammary cancer initiation in rats. Anticancer Res. 1988, 8, 113–117. [Google Scholar] [PubMed]
- Swanson, S.M.; Whitaker, L.M.; Stockard, C.R.; Myers, R.B.; Oelschlager, D.; Grizzle, W.E.; Juliana, M.M.; Grubbs, C.J. Hormone levels and mammary epithelial cell proliferation in rats treated with a regimen of estradiol and progesterone that mimics the preventive effect of pregnancy against mammary cancer. Anticancer Res. 1997, 17, 4639–4645. [Google Scholar] [PubMed]
- Sivaraman, L.; Stephens, L.C.; Markaverich, B.M.; Clark, J.A.; Krnacik, S.; Conneely, O.M.; O’Malley, B.W.; Medina, D. Hormone-induced refractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis 1998, 19, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, L.; Conneely, O.M.; Medina, D.; O’Malley, B.W. p53 is a potential mediator of pregnancy and hormone-induced resistance to mammary carcinogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 12379–12384. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, C.J.; Farnell, D.R.; Hill, D.L.; McDonough, K.C. Chemoprevention of N-nitroso-N-methylurea-induced mammary cancers by pretreatment with 17 beta-estradiol and progesterone. J. Natl. Cancer Inst. 1985, 74, 927–931. [Google Scholar] [PubMed]
- Russo, I.H.; Koszalka, M.; Russo, J. Human chorionic gonadotropin and rat mammary cancer prevention. J. Natl. Cancer Inst. 1990, 82, 1286–1289. [Google Scholar] [CrossRef]
- Bougaret, L.; Delort, L.; Billard, H.; Le Huede, C.; Boby, C.; De la Foye, A.; Rossary, A.; Mojallal, A.; Damour, O.; Auxenfans, C.; et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 2018, 13, e0191571. [Google Scholar] [CrossRef]
- Gyamfi, J.; Eom, M.; Koo, J.S.; Choi, J. Multifaceted Roles of Interleukin-6 in Adipocyte-Breast Cancer Cell Interaction. Transl. Oncol. 2018, 11, 275–285. [Google Scholar] [CrossRef]
- Clegg, D.J.; Brown, L.M.; Woods, S.C.; Benoit, S.C. Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes 2006, 55, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jo, K.J.; Kim, O.S.; Kim, B.J.; Kang, D.W.; Lee, K.H.; Baik, H.W.; Han, M.S.; Lee, S.K. Parenteral 17beta-estradiol decreases fasting blood glucose levels in non-obese mice with short-term ovariectomy. Life Sci. 2010, 87, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Wohlers, L.M.; Spangenburg, E.E. 17beta-estradiol supplementation attenuates ovariectomy-induced increases in ATGL signaling and reduced perilipin expression in visceral adipose tissue. J. Cell. Biochem. 2010, 110, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.; Gabbi, C.; Morani, A.; Warner, M.; Gustafsson, J.A. Participation of ERalpha and ERbeta in glucose homeostasis in skeletal muscle and white adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E124–E133. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Sharma, D. Metastasis suppression by adiponectin: LKB1 rises up to the challenge. Cell Adhes. Migr. 2010, 4, 358–362. [Google Scholar] [CrossRef]
- Mauro, L.; Naimo, G.D.; Gelsomino, L.; Malivindi, R.; Bruno, L.; Pellegrino, M.; Tarallo, R.; Memoli, D.; Weisz, A.; Panno, M.L.; et al. Uncoupling effects of estrogen receptor alpha on LKB1/AMPK interaction upon adiponectin exposure in breast cancer. FASEB J. 2018, 32, 4343–4355. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, F.; Razandi, M.; Pedram, A.; Harvey, B.J.; Levin, E.R. Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Mol. Endocrinol. 2012, 26, 2058–2070. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Ogawa, S.; Inoue, S.; Watanabe, T.; Hiroi, H.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M. The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem. Biophys. Res. Commun. 1998, 243, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, G.B.; Tremblay, A.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Labrie, F.; Giguere, V. Cloning, chromosomal localization, and functional analysis of the murine estrogen receptor beta. Mol. Endocrinol. 1997, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, F.; Pettersson, K.; Tujague, M.; Gustafsson, J.-Å. Functional Differences between the Amino-Terminal Domains of Estrogen Receptors alpha and beta. Mol. Pharmacol. 2000, 58, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rowan, B.G. Genes related to estrogen action in reproduction and breast cancer. Front. Biosci. 2005, 10, 2346–2372. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.; et al. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin. Cancer Res. 2008, 14, 5228–5235. [Google Scholar] [CrossRef]
- Gruvberger-Saal, S.K.; Bendahl, P.O.; Saal, L.H.; Laakso, M.; Hegardt, C.; Eden, P.; Peterson, C.; Malmstrom, P.; Isola, J.; Borg, A.; et al. Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin. Cancer Res. 2007, 13, 1987–1994. [Google Scholar] [CrossRef]
- Novelli, F.; Milella, M.; Melucci, E.; Di Benedetto, A.; Sperduti, I.; Perrone-Donnorso, R.; Perracchio, L.; Venturo, I.; Nistico, C.; Fabi, A.; et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: An observational prospective study. Breast Cancer Res. 2008, 10, R74. [Google Scholar] [CrossRef]
- Skliris, G.P.; Leygue, E.; Watson, P.H.; Murphy, L.C. Estrogen receptor alpha negative breast cancer patients: Estrogen receptor beta as a therapeutic target. J. Steroid Biochem. Mol. Biol. 2008, 109, 1–10. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Takubo, K.; Matsuura, M.; Ito, Y.; Akiyama, F.; Sakamoto, G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J. Clin. Oncol. 2008, 26, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Nakopoulou, L. The favourable prognostic value of oestrogen receptor immunohistochemical expression in breast cancer. J. Clin. Pathol. 2004, 57, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor beta in breast cancer. Endocr. Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Suman, V.J.; Subramaniam, M.; Wu, X.; Negron, V.; Gingery, A.; Pitel, K.S.; Shah, S.S.; Cunliffe, H.E.; McCullough, A.E.; et al. ERbeta1: Characterization, prognosis, and evaluation of treatment strategies in ERalpha-positive and -negative breast cancer. BMC Cancer 2014, 14, 749. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.C.; Frasor, J.; Komm, B.; Katzenellenbogen, B.S. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006, 147, 4831–4842. [Google Scholar] [CrossRef] [PubMed]
- Charn, T.H.; Liu, E.T.; Chang, E.C.; Lee, Y.K.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: Mutual restriction and competitive site selection. Mol. Endocrinol. 2010, 24, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Deroo, B.J.; Buensuceso, A.V. Minireview: Estrogen receptor-beta: Mechanistic insights from recent studies. Mol. Endocrinol. 2010, 24, 1703–1714. [Google Scholar] [CrossRef]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Wu, W.; Castaneda, J.; Krishnamurthy, S.; Webb, P.; Gustafsson, J.A.; Thomas, C. Somatic loss of estrogen receptor beta and p53 synergize to induce breast tumorigenesis. Breast Cancer Res. 2017, 19, 79. [Google Scholar] [CrossRef]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.A.; Thomas, C. ERbeta decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget 2016, 7, 13599–13611. [Google Scholar] [CrossRef]
- Jonsson, P.; Katchy, A.; Williams, C. Support of a bi-faceted role of estrogen receptor beta (ERbeta) in ERalpha-positive breast cancer cells. Endocr. Relat. Cancer 2014, 21, 143–160. [Google Scholar] [CrossRef]
- Naaz, A.; Zakroczymski, M.; Heine, P.; Taylor, J.; Saunders, P.; Lubahn, D.; Cooke, P.S. Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): A potential role for estrogen receptor beta (ERbeta). Horm. Metab. Res. 2002, 34, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Manente, A.G.; Valenti, D.; Pinton, G.; Jithesh, P.V.; Daga, A.; Rossi, L.; Gray, S.G.; O’Byrne, K.J.; Fennell, D.A.; Vacca, R.A.; et al. Estrogen receptor beta activation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo. Oncogenesis 2013, 2, e72. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Karthik, G.M.; Lovrot, J.; Haglund, F.; Rosin, G.; Katchy, A.; Zhang, X.; Viberg, L.; Frisell, J.; Williams, C.; et al. Estrogen Receptor beta as a Therapeutic Target in Breast Cancer Stem Cells. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef]
- Hainaut, P.; Hollstein, M. p53 and Human Cancer: The First Ten Thousand Mutations. Adv. Cancer Res. 1999, 77, 81–137. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic Enzymes Can Modulate Cellular Life Span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Kamp, W.M.; Wang, P.Y.; Hwang, P.M. TP53 mutation, mitochondria and cancer. Curr. Opin. Genet. Dev. 2016, 38, 16–22. [Google Scholar] [CrossRef]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef]
- Vahsen, N.; Cande, C.; Briere, J.J.; Benit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Y.; Jin, A.; Tikunov, A.P.; Zhou, L.; Tollini, L.A.; Leslie, P.; Kim, T.H.; Li, L.O.; Coleman, R.A.; et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, E2414–E2422. [Google Scholar] [CrossRef] [PubMed]
- Assaily, W.; Rubinger, D.A.; Wheaton, K.; Lin, Y.; Ma, W.; Xuan, W.; Brown-Endres, L.; Tsuchihara, K.; Mak, T.W.; Benchimol, S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 2011, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Yu, G.; Jiang, L.; Li, T.; Lin, Q.; Tang, Y.; Gu, W. p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle 2013, 12, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Ruiz-Lozano, P.; Hixon, M.L.; Wagner, M.W.; Flores, a.I.; Ikawa, S.; Baldwin, a.S.; Chien, K.R.; Gualberto, A. P53 Is a Transcriptional Activator of the Muscle-Specific Phosphoglycerate Mutase Gene and Contributes in Vivo To the Control of Its Cardiac Expression. Cell Growth Differ. 1999, 10, 295–306. [Google Scholar] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Gao, X.; Mei, Y.; Wu, M. A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 2013, 5, 147–150. [Google Scholar] [CrossRef]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef]
- Konduri, S.D.; Medisetty, R.; Liu, W.; Kaipparettu, B.A.; Srivastava, P.; Brauch, H.; Fritz, P.; Swetzig, W.M.; Gardner, A.E.; Khan, S.A.; et al. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. USA 2010, 107, 15081–15086. [Google Scholar] [CrossRef]
- Sayeed, A.; Konduri, S.D.; Liu, W.; Bansal, S.; Li, F.; Das, G.M. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: Implications for the regulation of apoptosis. Cancer Res. 2007, 67, 7746–7755. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.H.; Rundhaug, J.E.; Tian, J.; Cullinan-Ammann, N.; Lambertz, I.; Conti, C.J.; Fuchs-Young, R. Transcriptional regulation of estrogen receptor-alpha by p53 in human breast cancer cells. Cancer Res. 2009, 69, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.T.; Shin, H.; Westerling, T.; Liu, X.S.; Brown, M. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 18060–18065. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Katzenellenbogen, B.S. Estrogen Receptor-beta Modulation of the ERalpha-p53 Loop Regulating Gene Expression, Proliferation, and Apoptosis in Breast Cancer. Horm. Cancer 2017, 8, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 2011, 145, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, S.; Al Mulla, F.; Luqmani, Y.A. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS ONE 2011, 6, e20610. [Google Scholar] [CrossRef] [PubMed]
- Dip, R.; Lenz, S.; Gmuender, H.; Naegeli, H. Pleiotropic combinatorial transcriptomes of human breast cancer cells exposed to mixtures of dietary phytoestrogens. Food Chem. Toxicol. 2009, 47, 787–795. [Google Scholar] [CrossRef]
- Chang, E.C.; Charn, T.H.; Park, S.H.; Helferich, W.G.; Komm, B.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen Receptors alpha and beta as determinants of gene expression: Influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 2008, 22, 1032–1043. [Google Scholar] [CrossRef]
- Moggs, J.G.; Murphy, T.C.; Lim, F.L.; Moore, D.J.; Stuckey, R.; Antrobus, K.; Kimber, I.; Orphanides, G. Anti-proliferative effect of estrogen in breast cancer cells that re-express ERalpha is mediated by aberrant regulation of cell cycle genes. J. Mol. Endocrinol. 2005, 34, 535–551. [Google Scholar] [CrossRef]
- Sikora, M.J.; Cooper, K.L.; Bahreini, A.; Luthra, S.; Wang, G.; Chandran, U.R.; Davidson, N.E.; Dabbs, D.J.; Welm, A.L.; Oesterreich, S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014, 74, 1463–1474. [Google Scholar] [CrossRef]
- Stender, J.D.; Kim, K.; Charn, T.H.; Komm, B.; Chang, K.C.; Kraus, W.L.; Benner, C.; Glass, C.K.; Katzenellenbogen, B.S. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 2010, 30, 3943–3955. [Google Scholar] [CrossRef] [PubMed]
- Wardell, S.E.; Kazmin, D.; McDonnell, D.P. Research resource: Transcriptional profiling in a cellular model of breast cancer reveals functional and mechanistic differences between clinically relevant SERM and between SERM/estrogen complexes. Mol. Endocrinol. 2012, 26, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Bhat-Nakshatri, P.; Wang, G.; Appaiah, H.; Luktuke, N.; Carroll, J.S.; Geistlinger, T.R.; Brown, M.; Badve, S.; Liu, Y.; Nakshatri, H. AKT alters genome-wide estrogen receptor alpha binding and impacts estrogen signaling in breast cancer. Mol. Cell. Biol. 2008, 28, 7487–7503. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; Weaver, A.; Pradhan, M.; Dai, Y.; Miller, L.D.; Lin, C.Y.; Stanculescu, A. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009, 69, 8918–8925. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Nikolai, B.C.; Lanz, R.B.; York, B.; Dasgupta, S.; Mitsiades, N.; Creighton, C.J.; Tsimelzon, A.; Hilsenbeck, S.G.; Lonard, D.M.; Smith, C.L.; et al. HER2 Signaling Drives DNA Anabolism and Proliferation through SRC-3 Phosphorylation and E2F1-Regulated Genes. Cancer Res. 2016, 76, 1463–1475. [Google Scholar] [CrossRef]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Smith, R.A.; Maira, S.-M.; et al. ERα-Dependent E2F Transcription Can Mediate Resistance to Estrogen Deprivation in Human Breast Cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef]
- Gong, P.; Madak-Erdogan, Z.; Li, J.; Cheng, J.; Greenlief, C.M.; Helferich, W.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Transcriptomic analysis identifies gene networks regulated by estrogen receptor alpha (ERalpha) and ERbeta that control distinct effects of different botanical estrogens. Nucl. Recept. Signal. 2014, 12, e001. [Google Scholar] [CrossRef]
- Creighton, C.J.; Cordero, K.E.; Larios, J.M.; Miller, R.S.; Johnson, M.D.; Chinnaiyan, A.M.; Lippman, M.E.; Rae, J.M. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome Biol. 2006, 7, R28. [Google Scholar] [CrossRef]
- Coser, K.R.; Chesnes, J.; Hur, J.; Ray, S.; Isselbacher, K.J.; Shioda, T. Global analysis of ligand sensitivity of estrogen inducible and suppressible genes in MCF7/BUS breast cancer cells by DNA microarray. Proc. Natl. Acad. Sci. USA 2003, 100, 13994–13999. [Google Scholar] [CrossRef]
- Webster, K.A. Regulation of glycolytic enzyme RNA transcriptional rates by oxygen availability in skeletal muscle cells. Mol. Cell. Biochem. 1987, 77, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A.; Gunning, P.; Hardeman, E.; Wallace, D.C.; Kedes, L. Coordinate reciprocal trends in glycolytic and mitochondrial transcript accumulations during the in vitro differentiation of human myoblasts. J. Cell. Physiol. 1990, 142, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef]
- Davison, C.A.; Durbin, S.M.; Thau, M.R.; Zellmer, V.R.; Chapman, S.E.; Diener, J.; Wathen, C.; Leevy, W.M.; Schafer, Z.T. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res. 2013, 73, 3704–3715. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grunewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef]
- Morris, B.A.; Burkel, B.; Ponik, S.M.; Fan, J.; Condeelis, J.S.; Aguirre-Ghiso, J.A.; Castracane, J.; Denu, J.M.; Keely, P.J. Collagen Matrix Density Drives the Metabolic Shift in Breast Cancer Cells. EBioMedicine 2016, 13, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Svastova, E.; Hulikova, A.; Rafajova, M.; Zat’ovicova, M.; Gibadulinova, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zaguilan, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Shuster, S.; Neudecker, B.A.; Formby, B. Lactate stimulates fibroblast expression of hyaluronan and CD44: The Warburg effect revisited. Exp. Cell Res. 2002, 276, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Constant, J.S.; Feng, J.J.; Zabel, D.D.; Yuan, H.; Suh, D.Y.; Scheuenstuhl, H.; Hunt, T.K.; Hussain, M.Z. Lactate elicits vascular endothelial growth factor from macrophages: A possible alternative to hypoxia. Wound Repair Regen. 2000, 8, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Lewis, C.; Murdoch, C. Macrophage responses to hypoxia: Implications for tumor progression and anti-cancer therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Murata, Y.; Ohteki, T.; Koyasu, S.; Hamuro, J. IFN-γ and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur. J. Immunol. 2002, 32, 2866–2873. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1367. [Google Scholar] [CrossRef]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Tsirigos, A.; Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010, 9, 3485–3505. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Biol. Ther. 2011, 12, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Gazi, E.; Gardner, P.; Lockyer, N.P.; Hart, C.A.; Brown, M.D.; Clarke, N.W. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J. Lipid Res. 2007, 48, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef]
- Thol, F.; Weissinger, E.M.; Krauter, J.; Wagner, K.; Damm, F.; Wichmann, M.; Gohring, G.; Schumann, C.; Bug, G.; Ottmann, O.; et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica 2010, 95, 1668–1674. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Fathi, A.T.; Sadrzadeh, H.; Comander, A.H.; Higgins, M.J.; Bardia, A.; Perry, A.; Burke, M.; Silver, R.; Matulis, C.R.; Straley, K.S.; et al. Isocitrate dehydrogenase 1 (IDH1) mutation in breast adenocarcinoma is associated with elevated levels of serum and urine 2-hydroxyglutarate. Oncologist 2014, 19, 602–607. [Google Scholar] [CrossRef]
- Tang, X.; Lin, C.C.; Spasojevic, I.; Iversen, E.S.; Chi, J.T.; Marks, J.R. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014, 16, 415. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathe, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 2014, 124, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.A.; Gretz, D.M.; Taylor, P.C.; James, S.J.; Pogribny, I.P.; Miller, B.J.; Henning, S.M.; Swendseid, M.E. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J. Nutr. 1998, 128, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Rampersaud, G.C.; Kauwell, G.P.; Hutson, A.D.; Cerda, J.J.; Bailey, L.B. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 2000, 72, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.M.; Giuliano, A.R.; Piyathilake, C.; Nour, M.; Hatch, K. Hypomethylation in cervical tissue: Is there a correlation with folate status? Cancer Epidemiol. Biomark. Prev. 1998, 7, 901–906. [Google Scholar]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Yung, W.K.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef]
- Garcia, J.M.; Silva, J.; Pena, C.; Garcia, V.; Rodriguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; Espana, P.; Bonilla, F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef]
- Esteller, M.; Avizienyte, E.; Corn, P.G.; Lothe, R.A.; Baylin, S.B.; Aaltonen, L.A.; Herman, J.G. Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene 2000, 19, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Fitzgerald, M.P.; Venkataraman, S.; Vorrink, S.U.; Case, A.J.; Teoh, M.L.; Domann, F.E. Aberrant promoter CpG methylation is a mechanism for impaired PHD3 expression in a diverse set of malignant cells. PLoS ONE 2011, 6, e14617. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.; Zino, S.; Macintyre, A.; Kingsmore, D.; Payne, A.P.; George, W.D.; Shiels, P.G. Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 2006, 95, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Hsieh, Y.J.; Cheng, W.C.; Lin, C.P.; Lin, Y.S.; Yang, S.F.; Chen, C.C.; Izumiya, Y.; Yu, J.S.; Kung, H.J.; et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jin, N.; Sun, W.; Li, X.; Liu, B.; Xie, Z.; Qu, J.; Xu, J.; Yang, X.; Su, Y.; et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 2017, 36, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570. [Google Scholar] [CrossRef]
- Sullivan, L.B. Metabolic Frugality Marks Cancer Cells for Immune Targeting. Cell 2018, 174, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Delsite, R.; Kachhap, S.; Anbazhagan, R.; Gabrielson, E.; Singh, K.K. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol. Cancer 2002, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef] [PubMed]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bai, R.K.; Trieu, R.; Wong, L.J. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta 2010, 1797, 29–37. [Google Scholar] [CrossRef]
- Kaipparettu, B.A.; Ma, Y.; Wong, L.J. Functional effects of cancer mitochondria on energy metabolism and tumorigenesis: Utility of transmitochondrial cybrids. Ann. N. Y. Acad. Sci. 2010, 1201, 137–146. [Google Scholar] [CrossRef]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.J. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef]
- Guha, M.; Srinivasan, S.; Ruthel, G.; Kashina, A.K.; Carstens, R.P.; Mendoza, A.; Khanna, C.; Van Winkle, T.; Avadhani, N.G. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene 2014, 33, 5238–5250. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Ma, W.; Park, J.Y.; Celi, F.S.; Arena, R.; Choi, J.W.; Ali, Q.A.; Tripodi, D.J.; Zhuang, J.; Lago, C.U.; et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med. 2013, 368, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Zhuang, J.; Li, J.; Hwang, P.M. p53 as guardian of the mitochondrial genome. FEBS Lett. 2016, 590, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, H.J.; Wu, X.; Huo, L.; Kim, S.J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Invest. 2017, 127, 2091–2105. [Google Scholar] [CrossRef]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef]
- Furman, E.; Rushkin, E.; Margalit, R.; Bendel, P.; Degani, H. Tamoxifen induced changes in MCF7 human breast cancer: In vitro and in vivo studies using nuclear magnetic resonance spectroscopy and imaging. J. Steroid Biochem. Mol. Biol. 1992, 43, 189–195. [Google Scholar] [CrossRef]
- Neeman, M.; Degani, H. Metabolic studies of estrogen- and tamoxifen-treated human breast cancer cells by nuclear magnetic resonance spectroscopy. Cancer Res. 1989, 49, 589–594. [Google Scholar] [PubMed]
- Rivenzon-Segal, D.; Boldin-Adamsky, S.; Seger, D.; Seger, R.; Degani, H. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int. J. Cancer 2003, 107, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.H.; Paik, J.Y.; Jung, K.H.; Lee, K.H. 17beta-estradiol augments 18F-FDG uptake and glycolysis of T47D breast cancer cells via membrane-initiated rapid PI3K-Akt activation. J. Nucl. Med. 2010, 51, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Imbert-Fernandez, Y.; Clem, B.F.; O’Neal, J.; Kerr, D.A.; Spaulding, R.; Lanceta, L.; Clem, A.L.; Telang, S.; Chesney, J. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J. Biol. Chem. 2014, 289, 9440–9448. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjosne, H.; Giskeodegard, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Ding, K.; Chong, Q.Y.; Zhao, J.; Liu, Y.; Shao, Y.; Zhang, Y.; Yu, Q.; Xiong, Z.; Zhang, W.; et al. Post-transcriptional regulation of ERBB2 by miR26a/b and HuR confers resistance to tamoxifen in estrogen receptor-positive breast cancer cells. J. Biol. Chem. 2017, 292, 13551–13564. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1alpha Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285. [Google Scholar] [CrossRef]
- Radde, B.N.; Ivanova, M.M.; Mai, H.X.; Alizadeh-Rad, N.; Piell, K.; Van Hoose, P.; Cole, M.P.; Muluhngwi, P.; Kalbfleisch, T.S.; Rouchka, E.C.; et al. Nuclear respiratory factor-1 and bioenergetics in tamoxifen-resistant breast cancer cells. Exp. Cell Res. 2016, 347, 222–231. [Google Scholar] [CrossRef]
- Huber-Keener, K.J.; Liu, X.; Wang, Z.; Wang, Y.; Freeman, W.; Wu, S.; Planas-Silva, M.D.; Ren, X.; Cheng, Y.; Zhang, Y.; et al. Differential gene expression in tamoxifen-resistant breast cancer cells revealed by a new analytical model of RNA-Seq data. PLoS ONE 2012, 7, e41333. [Google Scholar] [CrossRef]
- Muluhngwi, P.; Alizadeh-Rad, N.; Vittitow, S.L.; Kalbfleisch, T.S.; Klinge, C.M. The miR-29 transcriptome in endocrine-sensitive and resistant breast cancer cells. Sci. Rep. 2017, 7, 5205. [Google Scholar] [CrossRef]
- Andrade-Vieira, R.; Goguen, D.; Bentley, H.A.; Bowen, C.V.; Marignani, P.A. Pre-clinical study of drug combinations that reduce breast cancer burden due to aberrant mTOR and metabolism promoted by LKB1 loss. Oncotarget 2014, 5, 12738–12752. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef]
- Thewes, V.; Simon, R.; Hlevnjak, M.; Schlotter, M.; Schroeter, P.; Schmidt, K.; Wu, Y.; Anzeneder, T.; Wang, W.; Windisch, P.; et al. The branched-chain amino acid transaminase 1 sustains growth of antiestrogen-resistant and ERalpha-negative breast cancer. Oncogene 2017, 36, 4124–4134. [Google Scholar] [CrossRef] [PubMed]
- Pitroda, S.P.; Khodarev, N.N.; Beckett, M.A.; Kufe, D.W.; Weichselbaum, R.R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 5837–5841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Chiu, L.L.; Sethi, S.K.; Koay, E.S. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell. Proteomics 2005, 4, 1686–1696. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.J.; Cook, R.S.; Manning, H.C.; Hicks, D.J.; Lafontant, A.; Arteaga, C.L.; Skala, M.C. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013, 73, 6164–6174. [Google Scholar] [CrossRef]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef]
- Tian, C.; Yuan, Z.; Xu, D.; Ding, P.; Wang, T.; Zhang, L.; Jiang, Z. Inhibition of glycolysis by a novel EGFR/HER2 inhibitor KU004 suppresses the growth of HER2+ cancer. Exp. Cell Res. 2017, 357, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, Y.; Koo, J.S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Hamilton, A.; Geng, S.; Hong, T.; Kalkum, M.; Momand, J.; Kane, S.E.; Huss, J.M. t-Darpp Activates IGF-1R Signaling to Regulate Glucose Metabolism in Trastuzumab-Resistant Breast Cancer Cells. Clin. Cancer Res. 2018, 24, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.I.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.G.; Crown, J.; Porter, R.K.; O’Driscoll, L. Neuromedin U alters bioenergetics and expands the cancer stem cell phenotype in HER2-positive breast cancer. Int. J. Cancer 2017, 140, 2771–2784. [Google Scholar] [CrossRef] [PubMed]
- Baumann, J.; Kokabee, M.; Wong, J.; Balasubramaniyam, R.; Sun, Y.; Conklin, D.S. Global metabolite profiling analysis of lipotoxicity in HER2/neu-positive breast cancer cells. Oncotarget 2018, 9, 27133–27150. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Wang, S.; Shiuan, E.; Brantley-Sieders, D.M.; Kim, L.C.; Reynolds, A.B.; Chen, J. The receptor tyrosine kinase EphA2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators YAP and TAZ. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Garcia-Castillo, V.; Lopez-Urrutia, E.; Villanueva-Sanchez, O.; Avila-Rodriguez, M.A.; Zentella-Dehesa, A.; Cortes-Gonzalez, C.; Lopez-Camarillo, C.; Jacobo-Herrera, N.J.; Perez-Plasencia, C. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J. Cancer 2017, 8, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the alpha-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Du, G.J.; Song, Z.H.; Lin, H.H.; Han, X.F.; Zhang, S.; Yang, Y.M. Luteolin as a glycolysis inhibitor offers superior efficacy and lesser toxicity of doxorubicin in breast cancer cells. Biochem. Biophys. Res. Commun. 2008, 372, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother. Pharmacol. 2016, 78, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chen, S.; Yang, W.; Cheng, X.; Ye, Y.; Mao, J.; Wu, X.; Huang, L.; Ji, J. FGFR4 Links Glucose Metabolism and Chemotherapy Resistance in Breast Cancer. Cell. Physiol. Biochem. 2018, 47, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Kanaan, Y.M.; Sampey, B.P.; Beyene, D.; Esnakula, A.K.; Naab, T.J.; Ricks-Santi, L.J.; Dasi, S.; Day, A.; Blackman, K.W.; Frederick, W.; et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genom. Proteom. 2014, 11, 279–294. [Google Scholar]
- Mohanti, B.K.; Rath, G.K.; Anantha, N.; Kannan, V.; Das, B.S.; Chandramouli, B.A.; Banerjee, A.K.; Das, S.; Jena, A.; Ravichandran, R.; et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: Phase I/II clinical trials on human cerebral gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 103–111. [Google Scholar] [CrossRef]
- Singh, D.; Banerji, A.K.; Dwarakanath, B.S.; Tripathi, R.P.; Gupta, J.P.; Mathew, T.L.; Ravindranath, T.; Jain, V. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther. Onkol. 2005, 181, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Dwarakanath, B.; Jain, V. Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Future Oncol. 2009, 5, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med 2016, 21, 373–380. [Google Scholar] [PubMed]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: Opportunities and challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
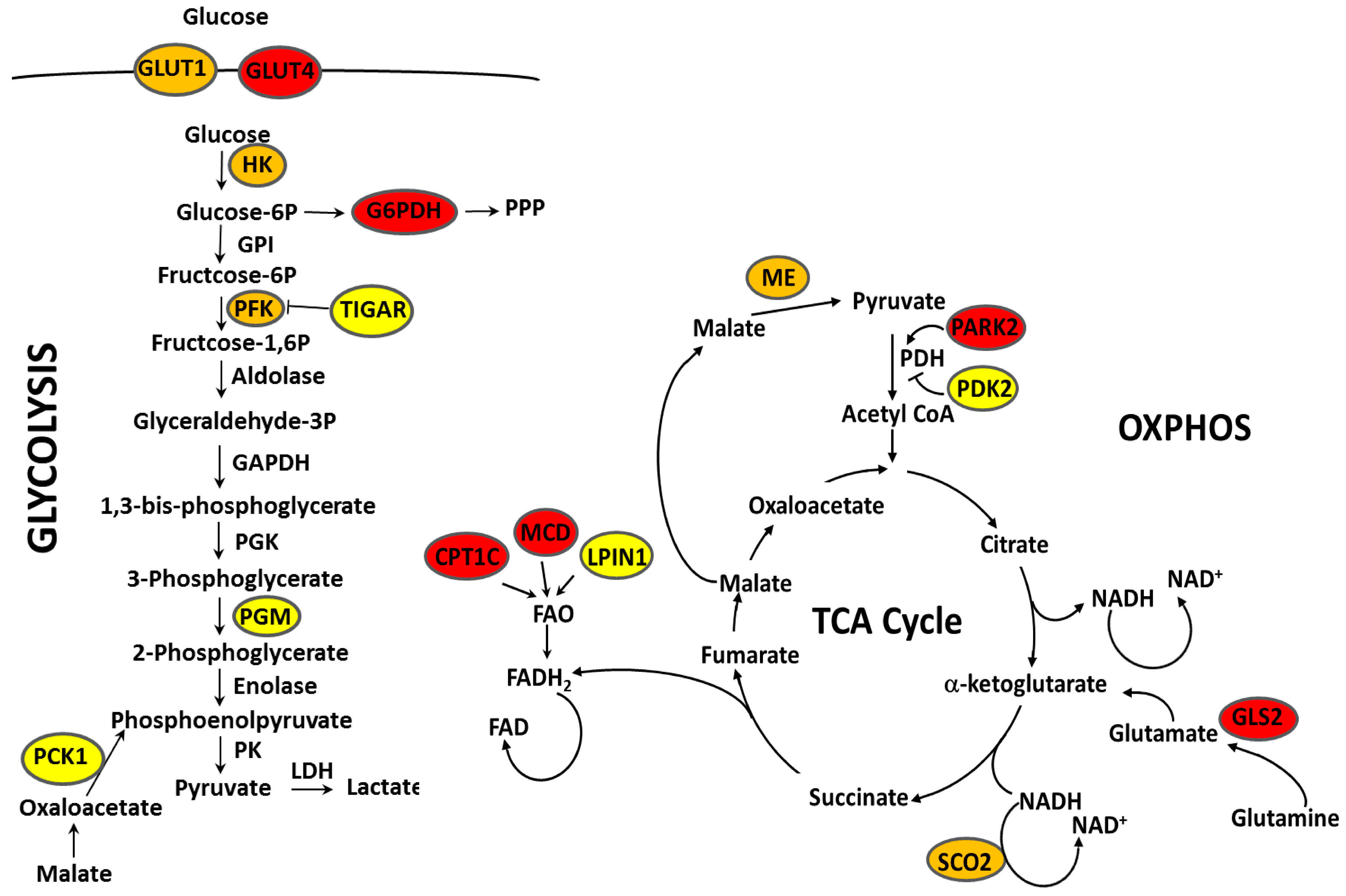
| Metabolic Targets of p53 | Regulation by p53 | p53 Status ∗ | Regulation by ERα | p53 Status ∗ | References |
|---|---|---|---|---|---|
| G6PD | Repression | wt | Activation | wt | [135,136] |
| SREBP1 | Repression | wt | Activation | wt | [136] |
| Repression | wt | [135] | |||
| PFK1(PFKM) | Repression | wt | Repression | wt | [135] |
| Activation | wt | [137] | |||
| PGM1 | Repression | wt | Repression | wt | [136,138] |
| PDK2 | Repression | wt | Repression | wt | [135] |
| PCK1 | Repression | wt | Activation | wt, R280K | [139,140] |
| HK2 | Repression/Activation | G103S, E256G | Repression | wt | [141,142] |
| Activation | wt | [136,138,143] | |||
| GLUT1(SLC2A1) | Repression | wt | Repression | wt | [136,142,144] |
| Activation | wt, R280K, E285K | [135,138,140,143,145,146,147] | |||
| GLUT4(SLC2A4) | Repression | wt | Activation | wt | [135,148] |
| ME1 | Repression | wt | Repression | wt, L194F, E285K | [135,138,149] |
| Activation | wt | [136,139,141] | |||
| ME2 | Repression | wt | Repression | wt | [136] |
| Activation | wt | [138] | |||
| TIGAR | Activation | wt | Activation | wt, L194F, E285K | [149] |
| PARK2 | Activation | wt | Repression | wt | [150] |
| SCO2 | Activation | wt | Activation | wt | [144] |
| Repression | wt | [138] | |||
| LPIN1 | Activation | wt | Activation | wt | [150] |
| CPT1C | Activation | wt | Repression | wt | [136] |
| GLS2 | Activation | wt | Repression | wt | [135] |
| MCD (MLYCD) | Activation | wt | Repression | wt | [136] |
| Target Protein and Pathway | Drug | Type | Indications | Clinical Trials |
|---|---|---|---|---|
| Glycolysis | ||||
| SGLT-2 | Dapagliflozin | Retrospective/Observational | Incidence of breast and bladder cancer | NCT02695121 |
| Hexokinase | 2-deoxy-D-glucose (2DG) | Phase I | Breast cancer and advanced solid malignancies | NCT00096707 |
| TCA Cycle and MRC | ||||
| PDK | Dichloroacetate | Phase II | Metastatic breast cancer or NSCLC | NCT01029925 |
| PDH/KGDH | CPI-613 | Phase II | Advanced solid tumors | NCT01832857 |
| ME-344 | Early Phase I | Her2- metastatic breast cancer | NCT02806817 | |
| Complex I | Metformin | Phase I/Phase II/Phase III | All breast cancer | multiple clinical trials |
| Lipid Synthesis | ||||
| FASN | TVB-2640 | Phase II | Her2+ metastatic breast cancer resistant to trastuzumab and taxanes | NCT03179904 |
| Omeprazole | Phase II | Triple negative breast cancer | NCT02595372 | |
| Conjugated Linoleic Acid (CLA) | Phase I | Metastatic breast cancer | NCT00908791 | |
| AA Metabolism | ||||
| Glutaminase | CB-839 | Phase I/Phase II | Advanced solid tumors/advanced TNBC | NCT02071862 NCT03057600 |
| Indoleamine 2,3 dioxygenase (IDO1) | Indoximod | Phase I/Phase II | Metastatic breast cancer | NCT01792050 NCT01042535 |
| Epacadostat | Phase I/Phase II | TNBC and other selected cancers | NCT02178722 | |
| Arginine deiminase (ADI) | ADI-PEG20 | Phase I | Her2- metastatic breast cancer | NCT01948843 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, N.; Das, G.M. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. https://doi.org/10.3390/cells8020089
Gandhi N, Das GM. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells. 2019; 8(2):89. https://doi.org/10.3390/cells8020089
Chicago/Turabian StyleGandhi, Nishant, and Gokul M Das. 2019. "Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications" Cells 8, no. 2: 89. https://doi.org/10.3390/cells8020089
APA StyleGandhi, N., & Das, G. M. (2019). Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells, 8(2), 89. https://doi.org/10.3390/cells8020089