Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment


Abstract
1. Introduction
2. Phenotype and Genetic Background
3. Mouse Models for HGPS
4. Classical Treatment Strategies for HGPS Progeria
5. Gene Therapy for HGPS Progeria
6. Gene Therapy Strategies Tested so far for Progeria
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonne, G. Laminopathies: Why make it simple when it can be complex? Neuromuscul. Disord. 2016, 26, S150–S151. [Google Scholar] [CrossRef]
- Maraldi, N.M.; Capanni, C.; Cenni, V.; Fini, M.; Lattanzi, G. Laminopathies and lamin-associated signaling pathways. J. Cell. Biochem. 2011, 112, 979–992. [Google Scholar] [CrossRef]
- Zaremba-Czogalla, M.; Dubinska-Magiera, M.; Rzepecki, R. Laminopathies: The molecular background of the disease and the prospects for its treatment. Cell. Mol. Biol. Lett. 2011, 16, 114–148. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Kazachkova, I.; Ten, S.; Garg, A. Severe Mandibuloacral Dysplasia-Associated Lipodystrophy and Progeria in a Young Girl with a Novel Homozygous Arg527Cys LMNA Mutation. J. Clin. Endocr. Metab. 2008, 93, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Muchir, A.; Recan, D.; Becane, H.M.; Urtizberea, J.A.; Penisson-Besnier, I.; Muntoni, F.; Merlini, L.; Toniolo, D.; Duboc, D.; et al. Spectrum of mutations in lamin A/C gene implicated in a new form of dilated cardiomyopathy with conduction defects and muscular dystrophy. Circulation 1999, 100, 617. [Google Scholar]
- Nishiuchi, S.; Makiyama, T.; Aiba, T.; Nakajima, K.; Hirose, S.; Kohjitani, H.; Yamamoto, Y.; Harita, T.; Hayano, M.; Wuriyanghai, Y.; et al. Gene-Based Risk Stratification for Cardiac Disorders in LMNA Mutation Carriers. Circ. Cardiovasc. Genet. 2017, 10, e001603. [Google Scholar] [CrossRef]
- Paquet, N.; Box, J.K.; Ashton, N.W.; Suraweera, A.; Croft, L.V.; Urquhart, A.J.; Bolderson, E.; Zhang, S.D.; O’Byrne, K.J.; Richard, D.J. Nestor-Guillermo Progeria Syndrome: A biochemical insight into Barrier-to-Autointegration Factor 1, alanine 12 threonine mutation. BMC Mol. Biol. 2014, 15, 27. [Google Scholar] [CrossRef]
- Cabanillas, R.; Cadinanos, J.; Villameytide, J.A.F.; Perez, M.; Longo, J.; Richard, J.M.; Alvarez, R.; Duran, N.S.; Illan, R.; Gonzalez, D.J.; et al. Nestor-Guillermo Progeria Syndrome: A Novel Premature Aging Condition With Early Onset and Chronic Development Caused by BANF1 Mutations. Am. J. Med. Genet. Part A 2011, 155, 2617–2625. [Google Scholar] [CrossRef]
- Columbaro, M.; Mattioli, E.; Schena, E.; Capanni, C.; Cenni, V.; Levy, N.; Navarro, C.L.; Del Coco, R.; Squarzoni, S.; Camozzi, D.; et al. Prelamin A processing and functional effects in restrictive dermopathy. Cell Cycle 2010, 9, 4766–4768. [Google Scholar] [CrossRef]
- Navarro, C.L.; De Sandre-Giovannoli, A.; Bernard, R.; Boccaccio, I.; Boyer, A.; Genevieve, D.; Hadj-Rabia, S.; Gaudy-Marqueste, C.; Smitt, H.S.; Vabres, P.; et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004, 13, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; del Campo, L.; Andres, V. Aging in the Cardiovascular System: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu. Rev. Physiol. 2018, 80, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Soria-Valles, C.; Carrero, D.; Gabau, E.; Velasco, G.; Quesada, V.; Barcena, C.; Moens, M.; Fieggen, K.; Mohrcken, S.; Owens, M.; et al. Novel LMNA mutations cause an aggressive atypical neonatal progeria without progerin accumulation. J. Med. Genet. 2016, 53, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, K.; Jiang, M.; Zhao, J. Hutchinson-Gilford progeria syndrome with scleroderma-like skin changes due to a homozygous missense LMNA mutation. J. Eur. Acad. Dermatol. 2016, 30, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Luo, N.; Hao, F.; Bai, Y. p.Pro4Arg mutation in LMNA gene: A new atypical progeria phenotype without metabolism abnormalities. Gene 2014, 546, 35–39. [Google Scholar] [CrossRef]
- Liang, L.L.; Zhang, H.W.; Gu, X.F. Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome: Evidence for autosomal recessive inheritance. Acta Paediatr. 2009, 98, 1365–1368. [Google Scholar] [CrossRef]
- Zirn, B.; Kress, W.; Grimm, T.; Berthold, L.D.; Neubauer, B.; Kuchelmeister, K.; Muller, U.; Hahn, A. Association of homozygous LMNA mutation R471C with new phenotype: Mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am. J. Med. Genet. Part A 2008, 146, 1049–1054. [Google Scholar] [CrossRef]
- Eriksson, M. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Levy, N.; Navarro, C.; Boccaccio, I.; Boyer, A.; Negre, P.; Devriendt, K.; Shafeghaty, Y.; Bridger, J.; Philip, N.; Bernard, R.; et al. Functional exploration of A type lamins and associated proteins in patients affected with Hutchinson-Gilford Progeria Syndrome caused by G608G mutation in LMNA. Am. J. Hum. Genet. 2003, 73, 554. [Google Scholar]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Isaev, N.K.; Genrikhs, E.E.; Oborina, M.V.; Stelmashook, E.V. Accelerated aging and aging process in the brain. Rev. Neurosci. 2018, 29, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Meinke, P.; Camozzi, D.; Mattioli, E.; Pfaeffle, R.; Siekmeyer, M.; Hirsch, W.; Horn, L.C.; Paasch, U.; Mitter, D.; et al. Progeroid laminopathy with restrictive dermopathy-like features caused by an isodisomic LMNA mutation p.R435C. Aging 2013, 5, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Thill, M.; Nguyen, T.D.; Wehnert, M.; Fischer, D.; Hausser, I.; Braun, S.; Jackisch, C. Restrictive dermopathy: A rare laminopathy. Arch. Gynecol. Obstet. 2008, 278, 201–208. [Google Scholar] [CrossRef]
- Dutta, A.K.; Danda, S. Restrictive Dermopathy. Pediatr. Neonatol. 2016, 57, 259. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005, 14, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Esteves-Vieira, V.; Courrier, S.; Boyer, A.; Nguyen, T.D.; Huong, L.T.T.; Meinke, P.; Schroder, W.; Cormier-Daire, V.; Sznajer, Y.; et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur. J. Hum. Genet. 2014, 22, 1002–1011. [Google Scholar] [CrossRef]
- Kirschner, J.; Brune, T.; Wehnert, M.; Denecke, J.; Wasner, C.; Feuer, A.; Marquardt, T.; Ketelsen, U.P.; Wieacker, P.; Bonnemann, C.G.; et al. p.S143F mutation in lamin A/C: A new phenotype combining myopathy and progeria. Ann. Neurol. 2005, 57, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.V.; Rottman, J.N.; Stewart, C.L. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. 2006, 14, 2167–2180. [Google Scholar] [CrossRef]
- Moulson, C.L.; Fong, L.G.; Gardner, J.M.; Farber, E.A.; Go, G.; Passariello, A.; Grange, D.K.; Young, S.G.; Miner, J.H. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum. Mutat. 2007, 28, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Reunert, J.; Wentzell, R.; Walter, M.; Jakubiczka, S.; Zenker, M.; Brune, T.; Rust, S.; Marquardt, T. Neonatal progeria: Increased ratio of progerin to lamin A leads to progeria of the newborn. Eur. J. Hum. Genet. 2012, 20, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Andres, D.A.; Spielmann, H.P.; Young, S.G.; Fong, L.G. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J. Clin. Investig. 2008, 118, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Chang, S.Y.; Ren, S.X.; Wang, Y.B.; Andres, D.A.; Spielmann, H.P.; Fong, L.G.; Young, S.G. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum. Mol. Genet. 2011, 20, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Schmidt, E.; Viceconte, N.; Strandgren, C.; Pernold, K.; Richard, T.J.C.; Van Leeuwen, F.W.; Dantuma, N.P.; Damberg, P.; Hultenby, K.; et al. Expression of progerin in aging mouse brains reveals structural nuclear abnormalities without detectible significant alterations in gene expression, hippocampal stem cells or behavior. Hum. Mol. Genet. 2015, 24, 1305–1321. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Davidovich, M.; Nissim-Rafinia, M.; Wiesel-Motiuk, N.; Bar, D.; Barkan, R.; Meshorer, E.; Gruenbaum, Y. Nuclear lamins: Key regulators of nuclear structure and activities. J. Cell. Mol. Med. 2009, 13, 1059–1085. [Google Scholar] [CrossRef] [PubMed]
- Butin-Israeli, V.; Adam, S.A.; Goldman, A.E.; Goldman, R.D. Nuclear lamin functions and disease. Trends Genet. 2012, 28, 464–471. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef]
- Dauer, W.T.; Worman, H.J. The nuclear envelope as a signaling node in development and disease. Dev. Cell 2009, 17, 626–638. [Google Scholar] [CrossRef]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J. Cell signaling abnormalities in cardiomyopathy caused by lamin A/C gene mutations. Biochem. Soc. Trans. 2018, 46, 37–42. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Abbas, A.; Irianto, J.; Ivanovska, I.L.; Xia, Y.T.; Tewari, M.; Discher, D.E. Progerin phosphorylation in interphase is lower and less mechanosensitive than lamin-A,C in iPS-derived mesenchymal stem cells. Nucleus 2018, 9, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Lai, J.K.; Xiong, Z.M.; Ren, M.; Moorer, M.C.; Stains, J.P.; Cao, K. Diminished Canonical beta-Catenin Signaling During Osteoblast Differentiation Contributes to Osteopenia in Progeria. J. Bone Miner. Res. 2018, 33, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Ivanovska, I.L.; Swift, J.; Discher, D.E. Lamin-A is Mechanosensitive to Matrix Stiffness and Couples to the Retinoic Acid Pathway in Differentiation. Biophys. J. 2014, 106, 8A. [Google Scholar] [CrossRef]
- Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [CrossRef]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior-life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef]
- Vidak, S.; Georgiou, K.; Fichtinger, P.; Naetar, N.; Dechat, T.; Foisner, R. Nucleoplasmic lamins define growth-regulating functions of lamina-associated polypeptide 2 alpha in progeria cells. J. Cell Sci. 2018, 131, jcs208462. [Google Scholar] [CrossRef] [PubMed]
- Zuela, N.; Dorfman, J.; Gruenbaum, Y. Global transcriptional changes caused by an EDMD mutation correlate to tissue specific disease phenotypes in C-elegans. Nucleus 2017, 8, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Barkan, R.; Gruenbaum, Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013, 12, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Machowska, M.; Piekarowicz, K.; Rzepecki, R. Regulation of lamin properties and functions: Does phosphorylation do it all? Open Biol. 2015, 5, 150094. [Google Scholar] [CrossRef] [PubMed]
- Melcon, G.; Kozlov, S.; Cutler, D.A.; Sullivan, T.; Hernandez, L.; Zhao, P.; Mitchell, S.; Nader, G.; Bakay, M.; Rottman, J.N.; et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006, 15, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Stubenvoll, A.; Rice, M.; Wietelmann, A.; Wheeler, M.; Braun, T. Attenuation of Wnt/beta-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum. Mol. Genet. 2015, 24, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Bonne, G.; Hayashi, Y.K.; Worman, H.J. Activation of MAPK in hearts of EMD null mice: Similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum. Mol. Genet. 2007, 16, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.A.; Collins, C.M.; Holaska, J.M. Rescue of emerin-null myogenic progenitor differentiation by inhibition of multiple MAP kinases or activation of HDAC3. Mol. Biol. Cell 2016, 27. [Google Scholar]
- Rajendran, V.; Purohit, R.; Sethumadhavan, R. In silico investigation of molecular mechanism of laminopathy caused by a point mutation (R482W) in lamin A/C protein. Amino Acids 2012, 43, 603–615. [Google Scholar] [CrossRef]
- Boschmann, M.; Engeli, S.; Moro, C.; Luedtke, A.; Adams, F.; Gorzelniak, K.; Rahn, G.; Mahler, A.; Dobberstein, K.; Kruger, A.; et al. LMNA Mutations, Skeletal Muscle Lipid Metabolism, and Insulin Resistance. J. Clin. Endocr. Metab. 2010, 95, 1634–1643. [Google Scholar] [CrossRef]
- Hernandez, L.; Roux, K.J.; Wong, E.S.; Mounkes, L.C.; Mutalif, R.; Navasankari, R.; Rai, B.; Cool, S.; Jeong, J.W.; Wang, H.; et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell 2010, 19, 413–425. [Google Scholar] [CrossRef]
- Wilson, K.L. Nuclear import pathway key to rescuing dominant progerin phenotypes. Sci. Signal. 2018, 11, eaat9448. [Google Scholar] [CrossRef] [PubMed]
- Bruston, F.; Delbarre, E.; Ostlund, C.; Worman, H.J.; Buendia, B.; Duband-Goulet, I. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 2010, 584, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Apte, K.; Stick, R.; Radmacher, M. Mechanics in human fibroblasts and progeria: Lamin A mutation E145K results in stiffening of nuclei. J. Mol. Recognit. 2017, 30, e2580. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.N.; Scaffidi, P.; Islam, M.F.; Yodh, A.G.; Wilson, K.L.; Misteli, T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 10271–10276. [Google Scholar] [CrossRef] [PubMed]
- Pekovic, V.; Gibbs-Seymour, I.; Markiewicz, E.; Alzoghaibi, F.; Benham, A.M.; Edwards, R.; Wenhert, M.; von Zglinicki, T.; Hutchison, C.J. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011, 10, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Pekovic, V.; Kill, I.; Benham, A.; Gruenbaum, Y.; Lattanzi, G.; von Zglinicki, T.; Hutchison, C. A Novel Role for Nucleoskeletal Protein Lamin a in Cellular Redox Homeostasis and Ageing: 258. Free Radic. Biol. Med. 2008, 45, S98. [Google Scholar]
- Mattioli, E.; Andrenacci, D.; Garofalo, C.; Prencipe, S.; Scotlandi, K.; Remondini, D.; Gentilini, D.; Di Blasio, A.M.; Valente, S.; Scarano, E.; et al. Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell 2018, 17, e12824. [Google Scholar] [CrossRef]
- Bridger, J.M.; Kill, I.R. Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosis. Exp. Gerontol. 2004, 39, 717–724. [Google Scholar] [CrossRef]
- Brunauer, R.; Kennedy, B.K. Progeria accelerates adult stem cell aging. Science 2015, 348, 1093–1094. [Google Scholar] [CrossRef]
- Dubinska-Magiera, M.; Zaremba-Czogalla, M.; Rzepecki, R. Muscle development, regeneration and laminopathies: How lamins or lamina-associated proteins can contribute to muscle development, regeneration and disease. Cell. Mol. Life Sci. 2013, 70, 2713–2741. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Cenni, V.; Lattanzi, G. Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br. J. Clin. Pharmacol. 2016, 82, 1229–1244. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andres-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; Lopez-Otin, C.; Andres, V. Vascular Smooth Muscle-Specific Progerin Expression Accelerates Atherosclerosis and Death in a Mouse Model of Hutchinson-Gilford Progeria Syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- Pfleghaar, K.B.; Taimen, P.; Butin-Israeli, V.; Shimi, T.; Langer-Freitag, S.; Markaki, Y.; Goldman, A.E.; Wehnert, M.; Goldman, R.D. Gene-rich chromosomal regions are preferentially localized in the lamin B deficient nuclear blebs of atypical progeria cells (vol 6, pg 66, 2015). Nucleus 2015, 6, 247. [Google Scholar] [CrossRef]
- Pekovic, V.; Harborth, J.; Broers, J.L.; Ramaekers, F.C.; van Engelen, B.; Lammens, M.; von Zglinicki, T.; Foisner, R.; Hutchison, C.; Markiewicz, E. Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 2007, 176, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ivorra, C.; Kubicek, M.; Gonzalez, J.M.; Sanz-Gonzalez, S.M.; Alvarez-Barrientos, A.; O’Connor, J.E.; Burke, B.; Andres, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Calvo, F.; Gonzalez, J.M.; Casar, B.; Andres, V.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef]
- Camozzi, D.; D’Apice, M.R.; Schena, E.; Cenni, V.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Squarzoni, S.; Ortolani, M.; Novelli, G.; et al. Altered chromatin organization and SUN2 localization in mandibuloacral dysplasia are rescued by drug treatment. Histochem. Cell Biol. 2012, 138, 643–651. [Google Scholar] [CrossRef]
- Cenni, V.; Capanni, C.; Mattioli, E.; Columbaro, M.; Wehnert, M.; Ortolani, M.; Fini, M.; Novelli, G.; Bertacchini, J.; Maraldi, N.M.; et al. Rapamycin treatment of Mandibuloacral Dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging 2014, 6, 755–769. [Google Scholar] [CrossRef]
- Columbaro, M.; Capanni, C.; Mattioli, E.; Novelli, G.; Parnaik, V.K.; Squarzoni, S.; Maraldi, N.M.; Lattanzi, G. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. Life Sci. 2005, 62, 2669–2678. [Google Scholar] [CrossRef]
- Kubben, N.; Adriaens, M.; Meuleman, W.; Voncken, J.W.; van Steensel, B.; Misteli, T. Mapping of lamin A- and progerin-interacting genome regions. Chromosoma 2012, 121, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Ishida, K.; Tsunoyama, T.A.; Toda, S.; Osoda, S.; Horigome, T.; Fisher, P.A.; Sugiyama, S. A-type and B-type lamins initiate layer assembly at distinct areas of the nuclear envelope in living cells. Exp. Cell Res. 2009, 315, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Delbarre, E.; Tramier, M.; Coppey-Moisan, M.; Gaillard, C.; Courvalin, J.C.; Buendia, B. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum. Mol. Genet. 2006, 15, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting Isoprenylcysteine Methylation Ameliorates Disease in a Mouse Model of Progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Voncken, J.W.; Demmers, J.; Calis, C.; van Almen, G.; Pinto, Y.; Misteli, T. Identification of differential protein interactors of lamin A and progerin. Nucleus 2010, 1, 513–525. [Google Scholar] [CrossRef]
- Zheng, X.B.; Hu, J.B.; Yue, S.B.; Kristiani, L.; Kim, M.; Sauria, M.; Taylor, J.; Kim, Y.; Zheng, Y.X. Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol. Cell 2018, 71, 802–815.e7. [Google Scholar] [CrossRef]
- Tang, Z.H.; Luo, O.J.; Li, X.W.; Zheng, M.Z.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef]
- Pendas, A.M. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Bergo, M.O. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef]
- Tonoyama, Y.; Shinya, M.; Toyoda, A.; Kitano, T.; Oga, A.; Nishimaki, T.; Katsumura, T.; Oota, H.; Wan, M.T.; Yip, B.W.P.; et al. Abnormal nuclear morphology is independent of longevity in a zmpste24-deficient fish model of Hutchinson-Gilford progeria syndrome (HGPS). Comp. Biochem. Phys. C 2018, 209, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Tu, Y.P.; Yang, S.H.; Tatar, A.; Nobumori, C.; Wu, D.; Young, S.G.; Fong, L.G. New Lmna knock-in mice provide a molecular mechanism for the ‘segmental aging’ in Hutchinson-Gilford progeria syndrome. Hum. Mol. Genet. 2014, 23, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Qiao, X.; Farber, E.; Chang, S.Y.; Fong, L.G.; Young, S.G. Eliminating the synthesis of mature lamin A reduces disease phenotypes in mice carrying a Hutchinson-Gilford progeria syndrome allele. J. Biol. Chem. 2008, 283, 7094–7099. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Bergo, M.O.; Toth, J.I.; Qiao, X.; Hu, Y.; Sandoval, S.; Meta, M.; Bendale, P.; Gelb, M.H.; Young, S.G.; et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 10291–10296. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Meta, M.; Qiao, X.; Frost, D.; Bauch, J.; Coffinier, C.; Majumdar, S.; Bergo, M.O.; Young, S.G.; Fong, L.G. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Investig. 2006, 116, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011, 2, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Zheng, Y.X. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem. Biophys. Res. Commun. 2013, 440, 8–13. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Cote, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Wang, Y.; Panteleyev, A.A.; Owens, D.M.; Djabali, K.; Stewart, C.L.; Worman, H.J. Epidermal expression of the truncated prelamin A causing Hutchinson-Gilford progeria syndrome: Effects on keratinocytes, hair and skin. Hum. Mol. Genet. 2008, 17, 2357–2369. [Google Scholar] [CrossRef]
- Sagelius, H.; Rosengardten, Y.; Hanif, M.; Erdos, M.R.; Rozell, B.; Collins, F.S.; Eriksson, M. Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J. Cell Sci. 2008, 121, 969–978. [Google Scholar] [CrossRef]
- Revechon, G.; Viceconte, N.; McKenna, T.; Sola-Carvajal, A.; Vrtacnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef] [PubMed]
- Gibbs-Seymour, I.; Markiewicz, E.; Bekker-Jensen, S.; Mailand, N.; Hutchison, C.J. Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell 2015, 14, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Lee, J.M.; Nobumori, C.; Tu, Y.P.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Larrieu, D.; Britton, S.; Demir, M.; Rodriguez, R.; Jackson, S.P. Chemical Inhibition of NAT10 Corrects Defects of Laminopathic. Cells Sci. 2014, 344, 527–532. [Google Scholar] [CrossRef]
- Balmus, G.; Larrieu, D.; Barros, A.C.; Collins, C.; Abrudan, M.; Demir, M.; Geisler, N.J.; Lelliott, C.J.; White, J.K.; Karp, N.A.; et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018, 9, 1700. [Google Scholar] [CrossRef]
- Ito, S.; Horikawa, S.; Suzuki, T.; Kawauchi, H.; Tanaka, Y.; Suzuki, T.; Suzuki, T. Human NAT10 Is an ATP-dependent RNA Acetyltransferase Responsible for N-4-Acetylcytidine Formation in 18 S Ribosomal RNA (rRNA). J. Biol. Chem. 2014, 289, 35724–35730. [Google Scholar] [CrossRef]
- Shen, Q.; Zheng, X.Z.; McNutt, M.A.; Guang, L.Z.; Sun, Y.; Wang, J.C.; Gong, Y.L.; Hou, L.; Zhang, B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 2009, 315, 1653–1667. [Google Scholar] [CrossRef]
- Kouzarides, T.; Vire, E.; Robson, S.; Breusegem, S.Y.; Kouzarides, T.; Jackson, S.P. Inhibition of the acetyltransferase NAT10 normalizes progeric and aging cells by rebalancing the Transportin-1 nuclear import pathway. Sci. Signal. 2018, 11, eaar5401. [Google Scholar] [CrossRef]
- Gabriel, D.; Shafry, D.D.; Gordon, L.B.; Djabali, K. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget 2017, 8, 64809–64826. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Columbaro, M.; Capanni, C.; D’Apice, M.R.; Cavallo, C.; Murdocca, M.; Lattanzi, G.; Squarzoni, S. All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 2015, 6, 29914–29928. [Google Scholar] [CrossRef]
- Wu, W.; Shan, J.A.; Bonne, G.; Worman, H.J.; Muchir, A. Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 632–638. [Google Scholar] [CrossRef]
- Matitioli, E.; Columbaro, M.; Capanni, C.; Santi, S.; Maraldi, N.M.; D’Apice, M.R.; Novelli, G.; Riccio, M.; Squarzoni, S.; Foisner, R.; et al. Drugs affecting prelamin A processing: Effects on heterochromatin organization. Exp. Cell Res. 2008, 314, 453–462. [Google Scholar] [CrossRef]
- Mallampalli, M.P.; Huyer, G.; Bendale, P.; Gelb, M.H.; Michaelis, S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 14416–14421. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Levy, N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; De Sandre-Giovannoli, A.; Levy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Michaelis, S. Permanently Farnesylated Prelamin A, Progeria, and Atherosclerosis. Circulation 2018, 138, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef]
- Ramanujulu, P.M.; Yang, T.M.; Yap, S.Q.; Wong, F.C.; Casey, P.J.; Wang, M.; Go, M.L. Functionalized indoleamines as potent, drug-like inhibitors of isoprenylcysteine carboxyl methyltransferase (Icmt). Eur. J. Med. Chem. 2013, 63, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Ly, D.H.; Lockhart, D.J.; Lerner, R.A.; Schultz, P.G. Mitotic misregulation and human aging. Science 2000, 287, 2486–2492. [Google Scholar] [CrossRef]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef]
- Fong, L.G.; Vickers, T.A.; Farber, E.A.; Choi, C.; Yun, U.J.; Hu, Y.; Yang, S.H.; Coffinier, C.; Lee, R.; Yin, L.; et al. Activating the synthesis of progerin, the mutant prelamin A in Hutchinson–Gilford progeria syndrome, with antisense oligonucleotides. Hum. Mol. Genet. 2009, 18, 2462–2471. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA-J. Am. Med. Assoc. 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical Trial of the Protein Farnesylation Inhibitors Lonafarnib, Pravastatin, and Zoledronic Acid in Children With Hutchinson-Gilford Progeria Syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W.; Collabora, P.C.T. Impact of Farnesylation Inhibitors on Survival in Hutchinson-Gilford Progeria Syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Good news in the nuclear envelope: Loss of lamin A might be a gain. J. Clin. Investig. 2006, 116, 632–634. [Google Scholar] [CrossRef]
- Fong, L.G. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2004, 101, 18111–18116. [Google Scholar] [CrossRef] [PubMed]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Levy, N.; De Sandre-Giovannoli, A. Antisense-Based Progerin Downregulation in HGPS-Like Patients’ Cells. Cells 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, E423–E431. [Google Scholar] [CrossRef] [PubMed]
- Sagelius, H.; Rosengardten, Y.; Schmidt, E.; Sonnabend, C.; Rozell, B.; Eriksson, M. Reversible phenotype in a mouse model of Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2008, 45, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Strandgren, C.; Nasser, H.A.; McKenna, T.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 2015, 29, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, J.; Freije, J.M.P.; Cabanillas, R.; Osorio, F.G.; Fraga, M.F.; Fernandez-Garcia, M.S.; Rad, R.; Fanjul, V.; Ugalde, A.P.; Liang, Q.; et al. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat. Commun. 2013, 4, 2268. [Google Scholar] [CrossRef]
- Gilbert, R.; Liu, A.B.; Petrof, B.J.; Nalbantoglu, J.; Karpati, G. Improved performance of a fully gutted adenovirus vector containing two full-length dystrophin cDNAs regulated by a strong promoter. Mol. Ther. 2002, 6, 501–509. [Google Scholar] [CrossRef]
- Liang, K.W.; Nishikawa, M.; Liu, F.; Sun, B.; Ye, Q.; Huang, L. Restoration of dystrophin expression in mdx mice by intravascular injection of naked DNA containing full-length dystrophin cDNA. Gene Ther. 2004, 11, 901–908. [Google Scholar] [CrossRef]
- Liu, M.; Yue, Y.; Harper, S.Q.; Grange, R.W.; Chamberlain, J.S.; Duan, D. Adeno-Associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 2005, 11, 245–256. [Google Scholar] [CrossRef]
- Herson, S.; Hentati, F.; Rigolet, A.; Behin, A.; Romero, N.B.; Leturcq, F.; Laforet, P.; Maisonobe, T.; Amouri, R.; Haddad, H.; et al. A phase I trial of adeno-associated virus serotype 1-gamma-sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C. Brain 2012, 135, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Rosales, X.Q.; Coley, B.D.; Galloway, G.; Lewis, S.; Malik, V.; Shilling, C.; Byrne, B.J.; Conlon, T.; et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010, 68, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted Gene Correction of Laminopathy-Associated LMNA Mutations in Patient-Specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.R.; Chen, L.S.; Libina, N.; Janes, J.; Martin, G.M.; Campisi, J.; Oshima, J. Correction of cellular phenotypes of Hutchinson-Gilford Progeria cells by RNA interference. Hum. Genet. 2005, 118, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Xiong, Z.M.; Cao, K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. USA 2014, 111, E2261–E2270. [Google Scholar] [CrossRef] [PubMed]
- Taskova, M.; Mantsiou, A.; Astakhova, K. Synthetic Nucleic Acid Analogues in Gene Therapy: An Update for Peptide-Oligonucleotide Conjugates. ChemBioChem 2017, 18, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.Y.; Muntoni, F.; Talbot, K.; et al. S Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef]
- Bao, T.L.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide development for the treatment of muscular dystrophies. Expert Opin. Orphan Drugs 2016, 4, 139–152. [Google Scholar] [CrossRef]



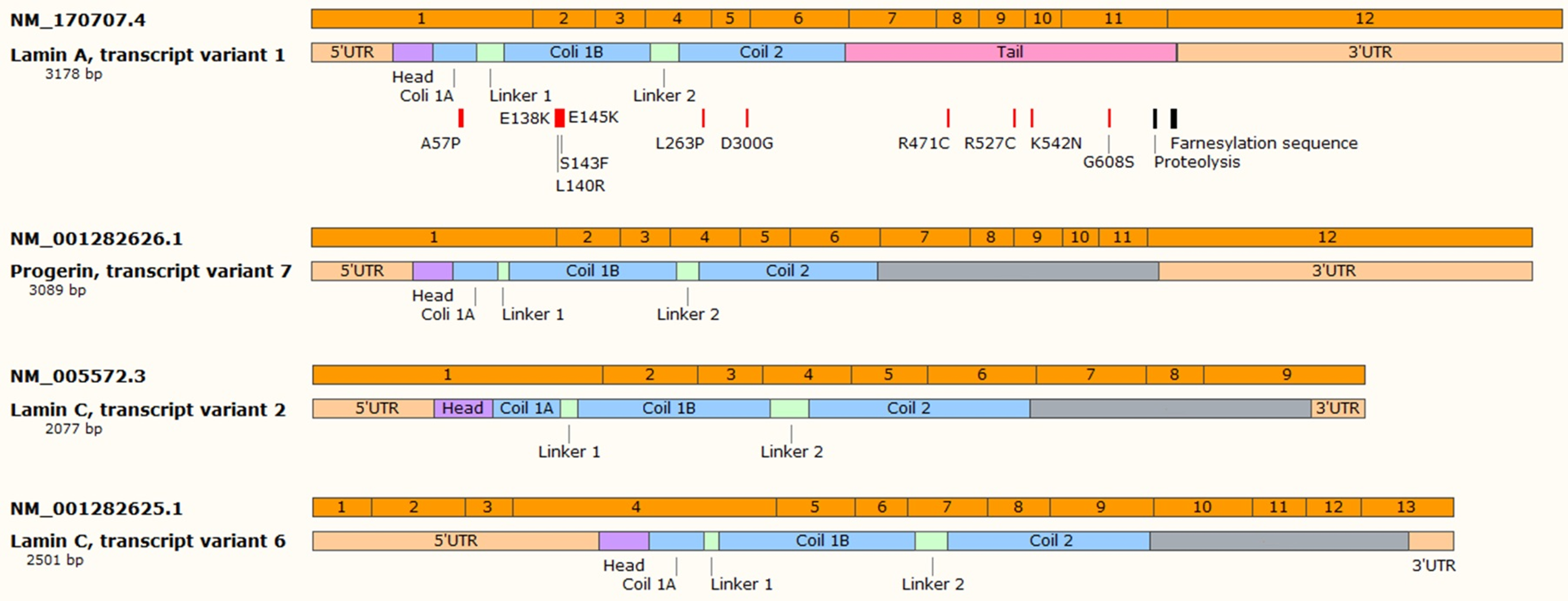{kind=link}
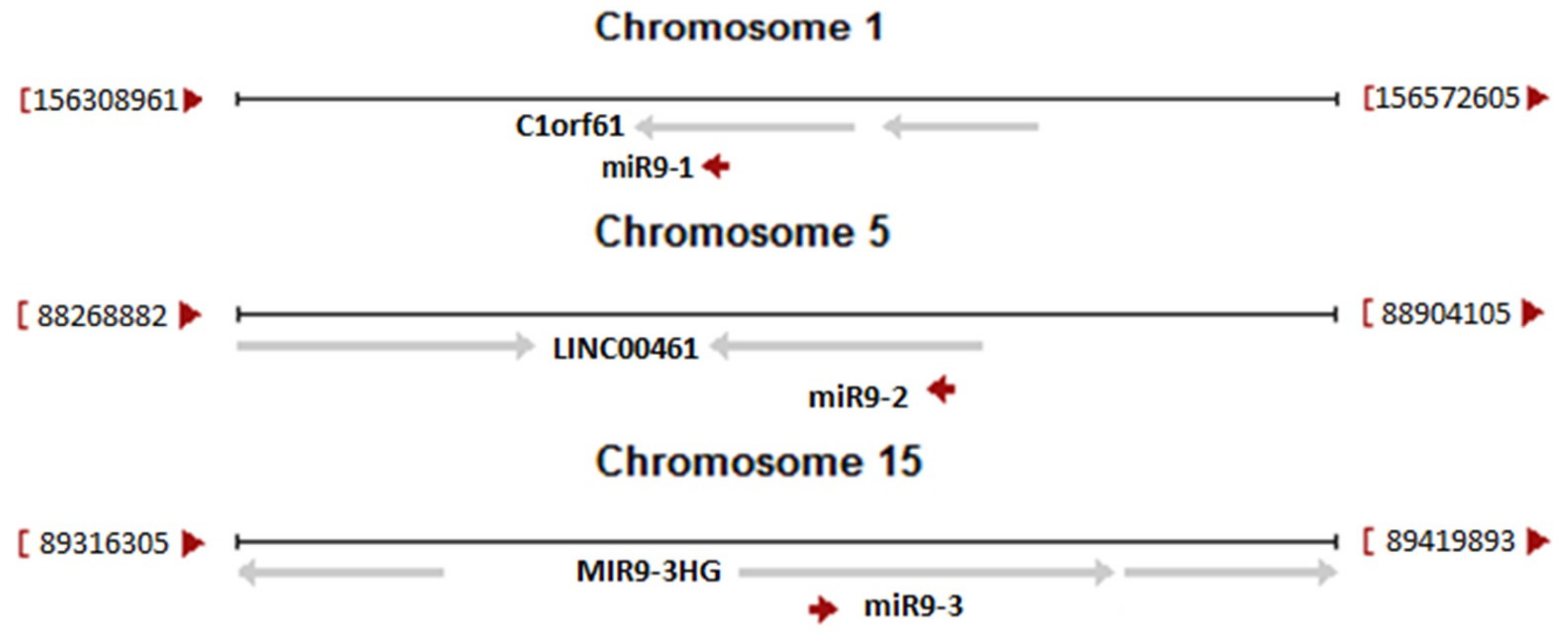{kind=link}
| Probe | Pre-miR-9-1 | Pre-miR-9-2 | Pre-miR-9-3 | miR-9-5p |
|---|---|---|---|---|
| Astrocyte-cerebral cortex, donor1 | 424 | 81 | 4 | 23,838 |
| Astrocyte-cerebral cortex, donor2 | 56 | 35 | 1 | 19,110 |
| Astrocyte-cerebral cortex, donor3 | 292 | 70 | 7 | 32,604 |
| Astrocyte-cerebellum, donor1 | 13 | 26 | 0 | 13,025 |
| Astrocyte-cerebellum, donor3 | 16 | 34 | 0 | 13,727 |
| Spinal cord, adult, donor10252 | 66 | 7 | 1 | 8,708 |
| Pineal gland, adult, donor10252 | 8 | 2 | 5 | 3,079 |
| Fibroblast-Mammary, donor1 | 0 | 0 | 0 | 0 |
| Fibroblast-Mammary, donor2 | 0 | 0 | 0 | 3 |
| Fibroblast-Mammary, donor3 | 0 | 0 | 0 | 914 |
| Prostate Epithelial Cells (polarized), donor1 | 0 | 0 | 0 | 848 |
| Smooth Muscle Cells-Brain Vascular, donor1 | 0 | 0 | 0 | 25 |
| Smooth Muscle Cells-Brain Vascular, donor2 | 0 | 5 | 0 | 582 |
| Smooth Muscle Cells-Brain Vascular, donor3 | 0 | 0 | 0 | 50 |
| Schwann Cells, donor1 | 0 | 1 | 0 | 378 |
| Strategy | Reference | Name and Target Sequence 5′->3′ | Target Transcript |
|---|---|---|---|
| Silencing of transcript variant 7, binds specifically to transcript coding for progerin | [144] | shRNA3 GGCTCAGGAGCCCAGAGCCCC | Variant 7 |
| [145] | |||
| [146] | |||
| Blocking the activated cryptic splice site in exon 11, binds directly to the sequence of cryptic splice site | [104] | Ex11 CTCAGGAGCCCAGGTGGGTGGACCC | Mutated pre-mRNA |
| [117] | |||
| [124] | |||
| [125] | |||
| [132] | |||
| Blocking the activated cryptic splice site in exon 11, binds upstream of this site | [124] | ASO 365 CTGTGCGGGACCTGCGGGCA | Mutated pre-mRNA |
| Binding to exon 10 splice site and shift splicing towards lamin C | [104] | Ex10 CCATCACCACCACGTGAGTGGTAGC | Pre-mRNA, mutated pre-mRNA |
| [117] | |||
| [132] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piekarowicz, K.; Machowska, M.; Dzianisava, V.; Rzepecki, R. Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment. Cells 2019, 8, 88. https://doi.org/10.3390/cells8020088
Piekarowicz K, Machowska M, Dzianisava V, Rzepecki R. Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment. Cells. 2019; 8(2):88. https://doi.org/10.3390/cells8020088
Chicago/Turabian StylePiekarowicz, Katarzyna, Magdalena Machowska, Volha Dzianisava, and Ryszard Rzepecki. 2019. "Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment" Cells 8, no. 2: 88. https://doi.org/10.3390/cells8020088
APA StylePiekarowicz, K., Machowska, M., Dzianisava, V., & Rzepecki, R. (2019). Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment. Cells, 8(2), 88. https://doi.org/10.3390/cells8020088