Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis

 ,
, 
Abstract

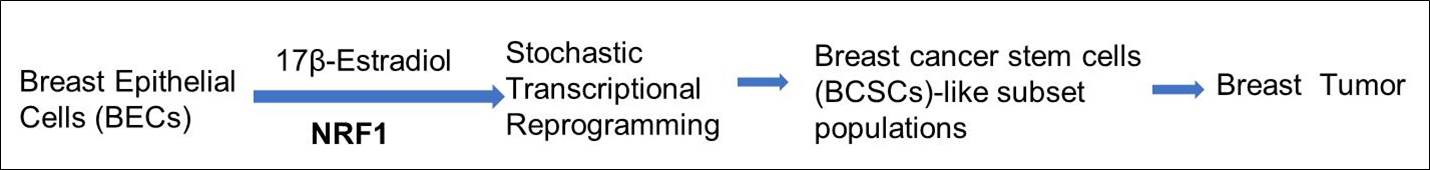
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies and Immunoblotting Analysis
2.3. Fluorescence-Activated Cell Sorting (FACS) and Immunofluorescence Detection
2.4. MTT, SRB, and BrdU Assays
2.5. FITC Annexin V Apoptosis Detection
2.6. Cell Invasion Assay
2.7. Colony Formation Assay
2.8. Tumorigenic Spheroid Assays
2.9. Cell Differentiation
2.10. Cell Migration Assay
2.11. Detection of Senescent Cells
2.12. Determination of Reactive Oxygen Species (ROS)
2.13. Drug-Resistant Characteristic of Mammospheres
2.14. In Vivo Tumorigenesis in Immunodeficient Mice
2.15. Chromatin Immunoprecipitation (ChIP) qPCR Assay to Analyze NRF-1 Binding to the Promoters of CXCR4 Genes
2.16. Luciferase Reporter Assay for Active CXCR4 Gene Promoter
2.17. Real-Time qRT-PCR Analysis for Detection of CXCR4 mRNA Levels
2.18. Immunofluorescence Study for CXCR4, 8-oxo-dG, and Real-Time qRT–PCR Analysis for CXCR4mRNA with Treatment of ROS Scavengers
2.19. Statistical Analyses
3. Results
3.1. Normal Breast Epithelial Cells Acquire Breast Tumor Initiating Properties by a Gain in NRF1 Activity in Conjunction with Exposure to E2
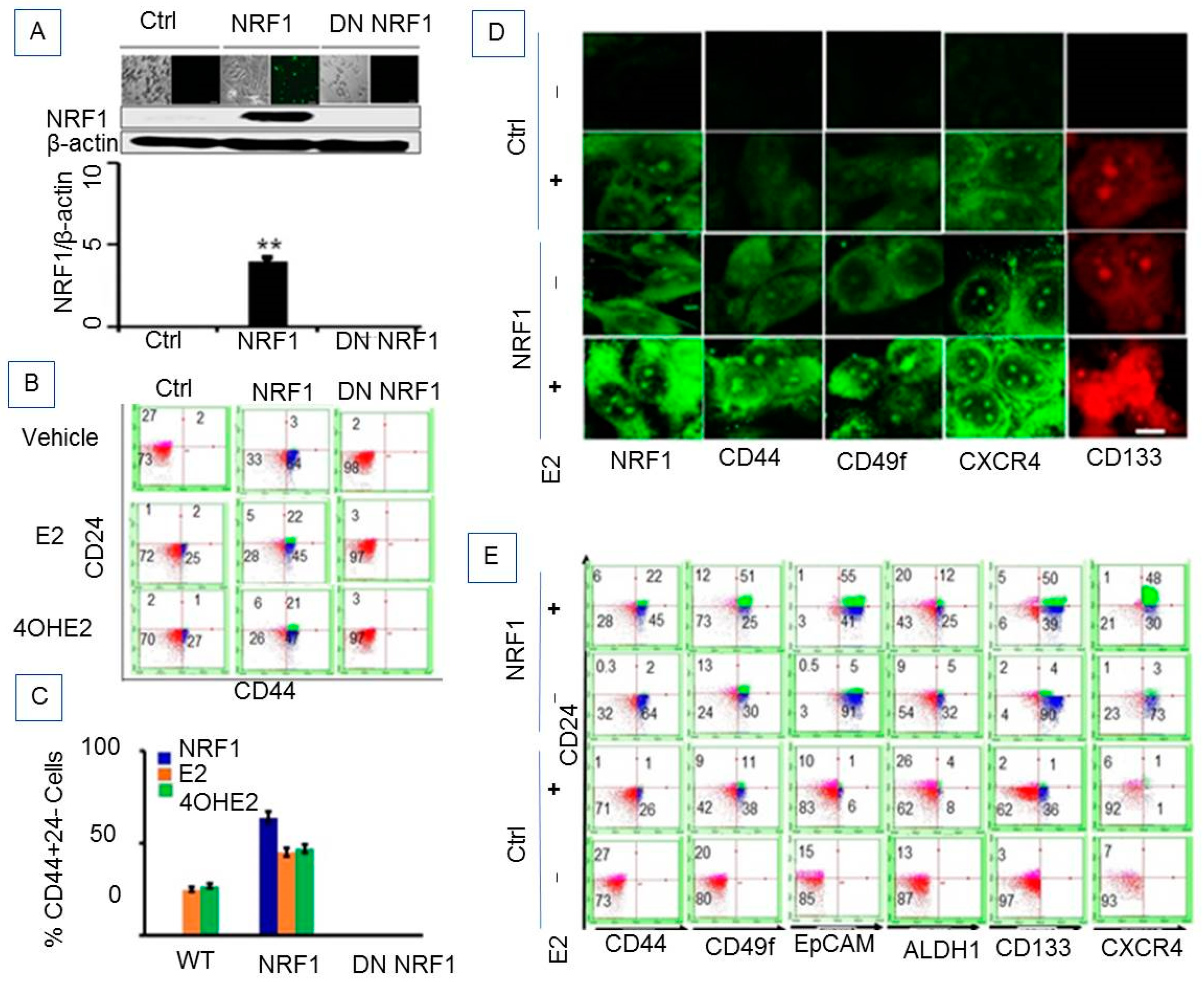
3.1.1. E2 Treatment Enhanced Breast Tumor-Initiating (Breast Cancer Stem) Cell Features
3.1.2. Ectopic NRF1 Expression Enriched the CD44 (High)/CD24 (Low) Progenitor Subtype in Mammary Epithelial Cells and Increased E2-Induced Progenitor Subpopulations
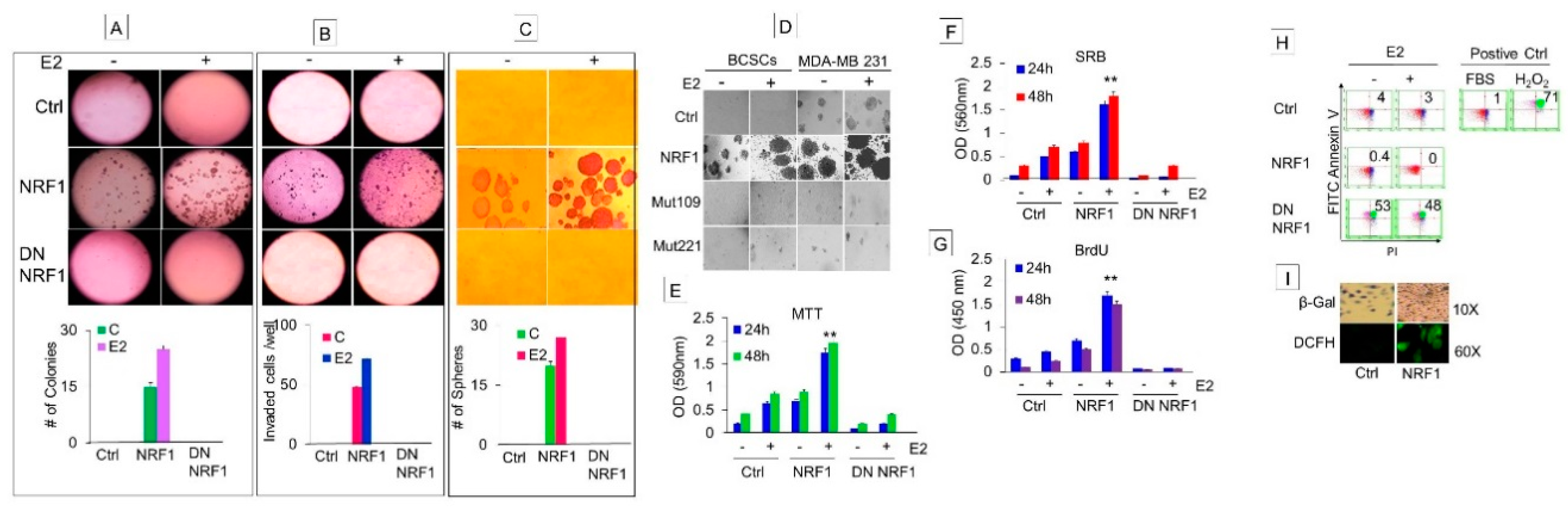
3.1.3. Phenotypic Characteristics of Estrogen-Induced Tumor Initiating Breast Cancer Cells
3.1.4. NRF1 Supports Self-Renewal of Breast Cancer Stem Cells
3.1.5. NRF1 and/or E2 Treatment Contributed to the Stochastic Re-Programming of Normal MCF10A Cells into Multiple Lineages of Human Breast Cancer Stem/Progenitor Cells
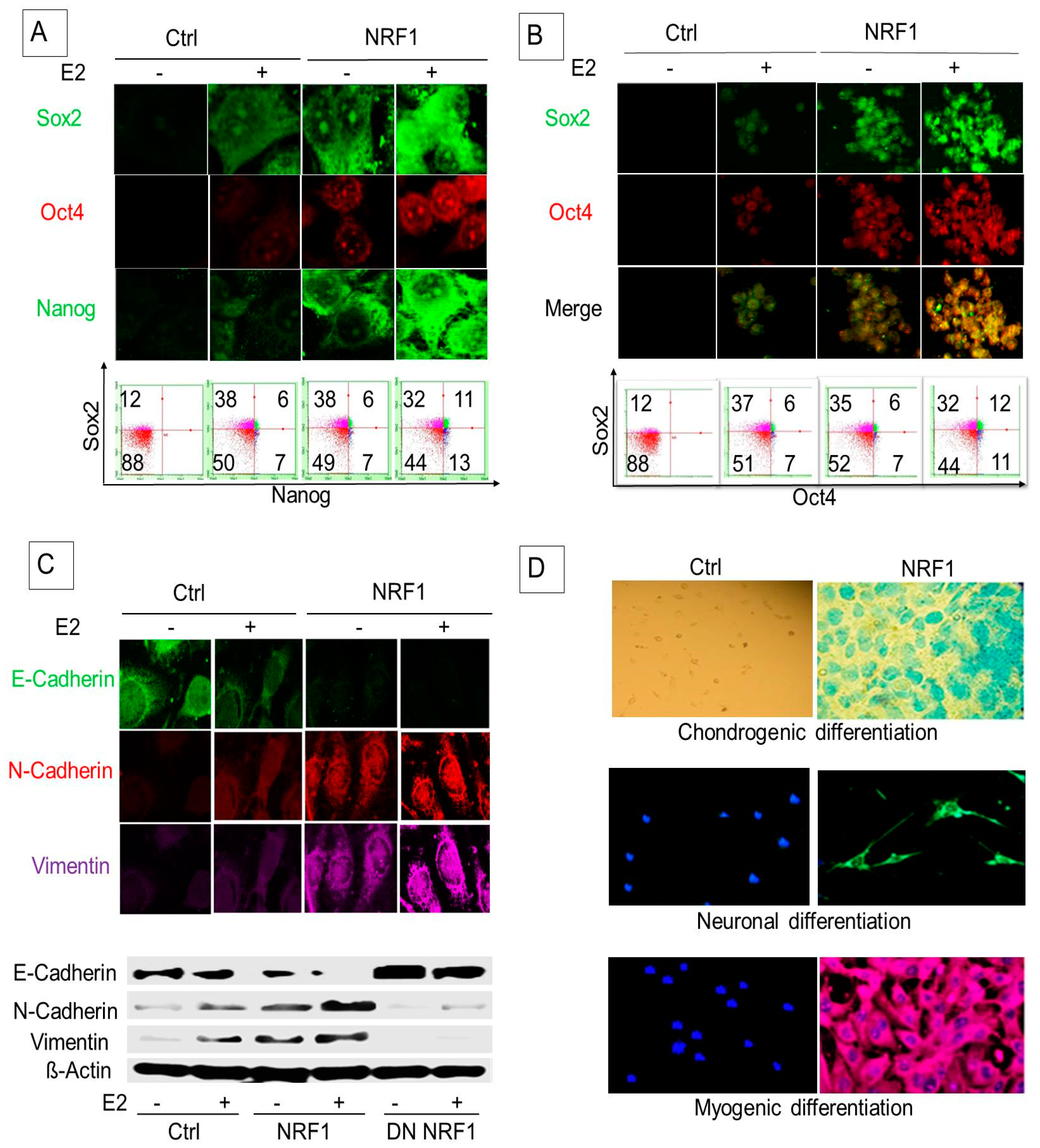
3.2. NRF1 and/or E2 Reprogramming Contributed to the Overexpression of Pluripotency Markers OCT4, NANOG, and SOX2
3.3. NRF1 Reprogramming Contributes to a Mesenchymal Phenotype Possessing Multi-Lineage Differentiation Potentials
3.4. Identification of the Mesenchymal-Like “Migrating and Metastasis Initiating” Breast Cancer Stem Cell Phenotype Clone
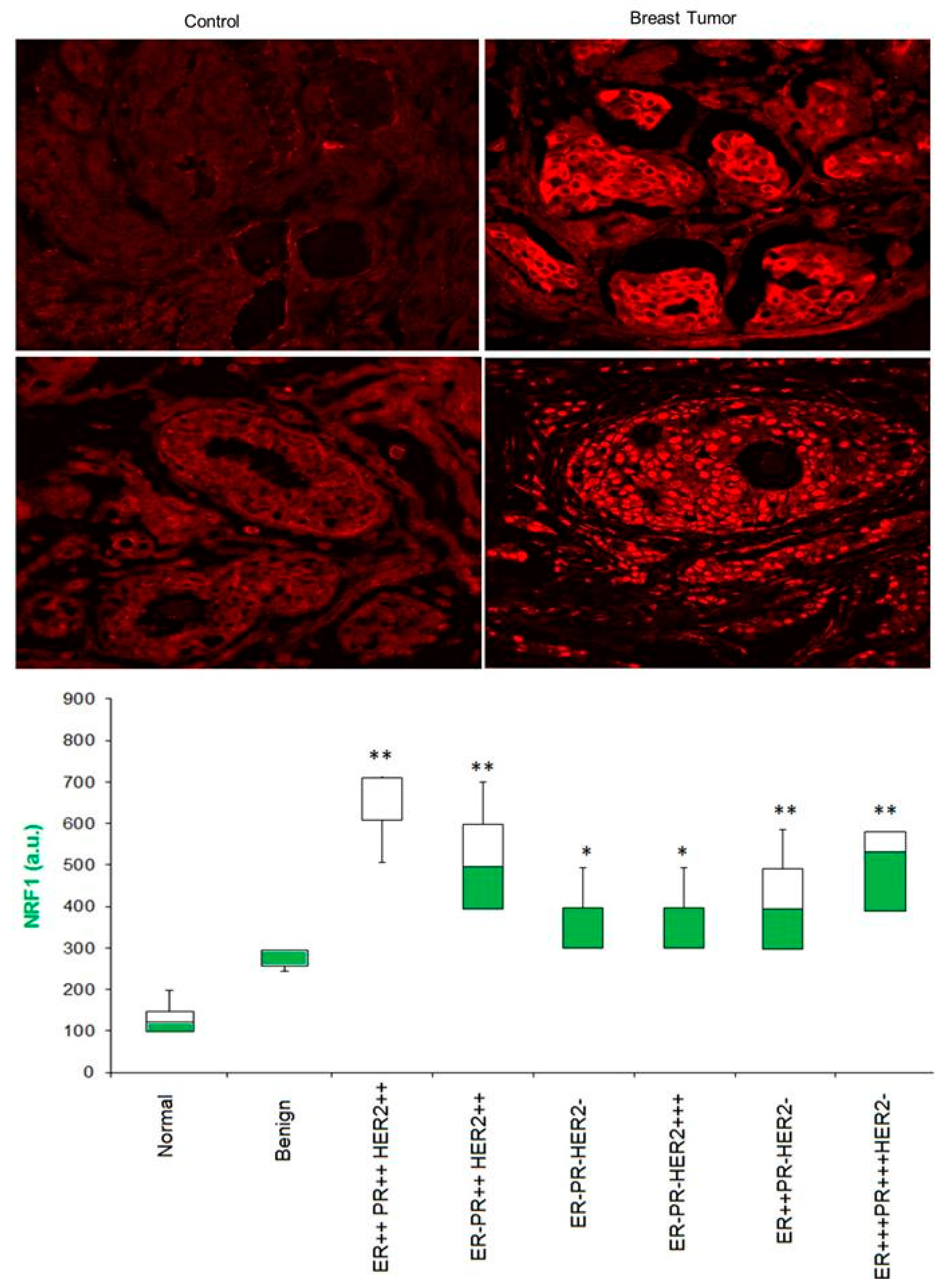
3.5. Is NRF1 Protein Enriched in Human Breast Tumor Specimens?
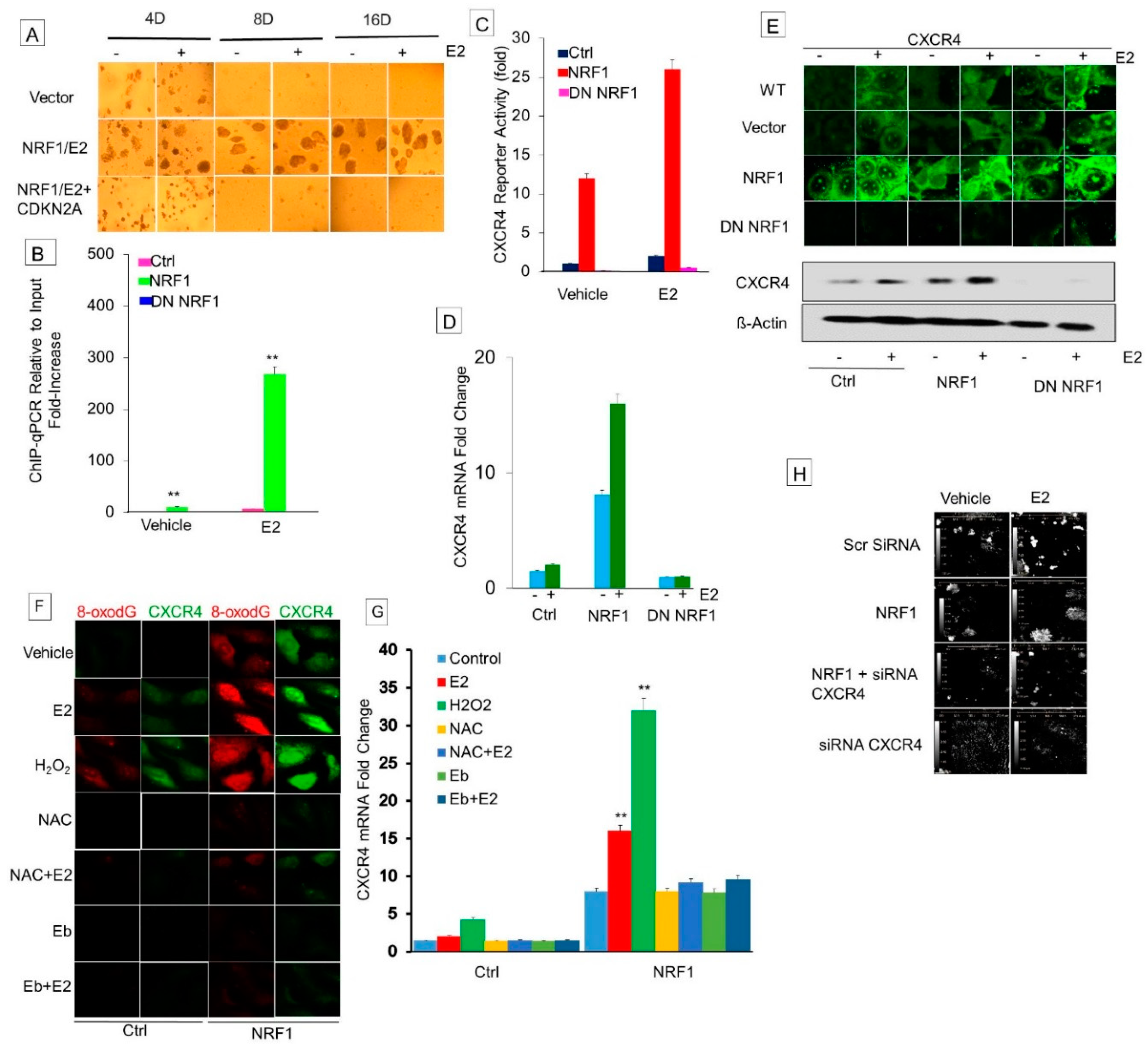
3.6. NRF1 Drives Breast Tumorigenesis through Regulating CXCR4 Signaling
3.6.1. Effect of Estrogen and NRF1 on CXCR4 Transcription
3.6.2. Determine Whether DNA Oxidation Is Necessary for NRF1-Mediated Transcriptional Activation of the CXCR4 Gene
3.6.3. Inhibition of Xenograft Tumor Growth of NRF1 BTICs by Silencing the Expression of CXCR4
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Elkon, R.; Linhart, C.; Sharan, R.; Shamir, R.; Shiloh, Y. Genome-wide In Silco identification of transcription regulators controlling the cell cycle in human cells. Genome Res. 2003, 13, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Balciunaite, E.; Blais, A.; Spektor, A.; Scarpulla, R.C.; Young, R.; Kluger, Y.; Dynlacht, B.D. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 2004, 16, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kawana, N.; Yamamoto, Y. Pathway analysis of ChIP-Seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Biol. 2013, 7, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Felty, Q.; Parkash, J.; Poppiti, R.; Roy, D. D Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS ONE 2013, 8, e54206. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Garba, N.A.; Penney, R.B.; Das, J.; Deoraj, A.; Singh, K.P.; Sarkar, S.; Felty, Q.; Yoo, C.; Jackson, R.M.; et al. Redox signalling to nuclear regulatory proteins by reactive oxygen species contributes to oestrogen-induced growth of breast cancer cells. Br. J. Cancer 2015, 112, 1687–1702. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.; Felty, Q.; Trevino, F.; Roy, D. Oncomine meta-analysis of breast cancer microarray data identifies upregulation of NRF-1 expression in human breast carcinoma. In Proceedings of the 18th World IMACS/MODSIM Congress, Cairns, Australia, 13–17 July 2009. [Google Scholar]
- Falco, M.M.; Bleda, M.; Carbonell-Caballero, J.; Dopazo, J. The pan-cancer pathological regulatory landscape. Sci. Rep. 2016, 6, 39709. [Google Scholar] [CrossRef] [PubMed]
- Travis, R.C.; Key, T.J. Oestrogen exposure and breast cancer risk. Breast Cancer Res. 2003, 5, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Smith, A.D.; Imoto, S.; Tsutsumi, S.; Aburatani, H.; Zhang, M.Q.; Akiyama, T. Integrative bioinformatics analysis of transcriptional regulatory programs in breast cancer cells. BMC Bioinf. 2008, 9, 404. [Google Scholar] [CrossRef] [PubMed]
- Bhawe, K.; Roy, D. Interplay between NRF1, E2F4 and MYC transcription factors regulating common target genes contributes to cancer development and progression. Cell. Oncol. 2018, 41, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Pogash, T.J.; Nguyen, T.D.; Russo, J. Development and characterization of two human triple-negative breast cancer cell lines with highly tumorigenic and metastatic capabilities. Cancer Med. 2016, 5, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Sobolika, T.; Sua, Y.; Wells, S. CXCR4 drives the metastatic phenotype in breast cancer through induction of CXCR2 and activation of MEK and PI3K pathways. Mol. Biol. Cell. 2014, 25, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.Q.; Zhang, S.; Li, S. BAG3 promotes stem cell-like phenotype in breast cancer by upregulation of CXCR4 via interaction with its transcript. Cell Death Dis. 2017, 8, e2933. [Google Scholar] [CrossRef] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.V.; Phan, N.L.C. What are markers for breast cancer stem cells? Prog. Stem Cells 2016, 3, 65–72. [Google Scholar] [CrossRef]
- Nishi, M.; Sakai, Y.; Akutsu, H.; Nagashima, Y.; Quinn, G.; Masui, S.; Kimura, H.; Perrem, K.; Umezawa, A.; Yamamoto, N.; et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene 2014, 33, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Talbot, L.J.; Bhattacharya, S.D.; Kuo, P.C. Epithelial-mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. Int. J. Biochem. Mol. Biol. 2012, 3, 117–136. [Google Scholar] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2105, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Chen, Z.; Zheng, R.; Cheng, Z.; Gong, X.; Wang, D. Clinicopathological significance of CD133 and CD44 expression in infiltrating ductal carcinoma and their relationship to angiogenesis. World J. Surg. Oncol. 2015, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Subik, K.; Lee, J.F.; Baxter, L.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; Tang, P. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wu, M.; Wang, N.; Zhang, Y.; Hua, J.; Tang, G.; Wang, Y. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 predicts a poor clinical outcome of breast cancer. Oncol. Lett. 2018, 15, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Ablett, M.P.; O’Brien, C.S.; Sims, A.H.; Farnie, G.; Clarke, R.B. A differential role for CXCR4 in the regulation of normal versus malignant breast stem cell activity. Oncotarget 2014, 5, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Short, S.P.; Neel, N.F.; Salvo, V.A.; Zhu, Y.; Elliott, S.; Wei, Y.; Yu, D.; Sun, M.; Muir, S.E.; et al. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011, 71, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Boudot, A.; Kerdivel, G.; Habauzit, D.; Eeckhoute, J.; Le Dily, F.; Flouriot, G.; Samson, M.; Pakdel, F. Differential Estrogen-Regulation of CXCL12 Chemokine Receptors, CXCR4 and CXCR7, Contributes to the Growth Effect of Estrogens in Breast Cancer Cells. PLoS ONE 2011, 6, e20898. [Google Scholar] [CrossRef] [PubMed]
- Wegner, S.A.; Ehrenberg, P.K.; Chang, G.; Dayhoff, D.E.; Sleeker, A.L.; Michael, N.L. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J. Biol Chem. 1998, 273, 4754–4760. [Google Scholar] [CrossRef] [PubMed]
- Angarica, V.E.; Sol, A. Modeling heterogeneity in the pluripotent state: A promising strategy for improving the efficiency and fidelity of stem cell differentiation. Bioessays 2016, 38, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Preciados, M.; Yoo, C.; Roy, D. Estrogenic endocrine disrupting chemicals influencing NRF11 regulated gene networks in the development of complex human brain diseases. Int. J. Mol. Sci. 2016, 17, 2086. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.; Das, J.; Felty, Q.; Yoo, C.; Poppiti, R.; Murrell, D.; Foster, P.J.; Roy, D. NRF1 motif sequence-enriched genes involved in ER/PR -ve Her2 +ve breast cancer signaling pathways. Breast Cancer Res. Treat. 2018, 172, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Benner, C.; Konovalov, S.; Mackintosh, C.; Hutt, K.R.; Stunnenberg, R.; Garcia-Bassets, I. Decoding a signature-based model of transcription cofactor recruitment dictated by cardinal cis-regulatory elements in proximal promoter regions. PLoS Genet. 2013, 9, e1003906. [Google Scholar] [CrossRef] [PubMed]
- Askarian-Amiri, M.E.; Seyfoddin, V.; Smart, C.E.; Wang, J.; Kim, J.E.; Hansji, H.; Baguley, B.C.; Finlay, G.J.; Leung, E.Y. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PLoS ONE 2014, 9, e102140. [Google Scholar] [CrossRef] [PubMed]
- Shahryari, A.; Jazi, M.S.; Samaei, N.M.; Mowla, S.J. Long non-coding RNA SOX2OT: Expression signature, splicing patterns, and emerging roles in pluripotency and tumorigenesis. Front. Genet. 2105, 6, 196. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.; Roy, D.; Das, J. Estrogen and nuclear respiratory factor 1 act as joint mediators of redox modulation and stem cell aging that contribute in the pathogenesis of breast cancer. Cancer Res. 2016, 76 (Suppl. 14), 3322. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Quan, D.; Zhang, F.; Zhang, H.; Xiao, T.; Hou, S.; Qiao, H.; Harismendy, O.; Wang, J.Y.; et al. Nuclear respiratory factor 1 promotes spheroid survival and mesenchymal transition in mammary epithelial cells. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Xue, Y.; Song, C.; Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2013, 110, 11493–11498. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 2006, 281, 324–333. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen Markers | V | E2 | NRF1 | NRF1+E2 | DN NRF1 | DN NRF1+E2 (%) |
|---|---|---|---|---|---|---|
| CD24+ | 27.46 | 01.46 | 00.26 | 05.36 | 02.56 | 02.72 |
| CD24−CD44- | 72.54 | 71.02 | 33.12 | 27.54 | 97.46 | 97.28 |
| CD24+CD44+ | ND | 01.46 | 02.36 | 21.54 | 0.0 | 0.0 |
| CD24−CD44+ | ND | 26.02 | 64.12 | 44.54 | 0.0 | 0.0 |
| CD24+CD44+CD49f+ | ND | 1.36 | 2.36 | 27.88 | 0.0 | 0.0 |
| CD24+CD44+CD49f- | ND | 10.00 | 31.00 | 29.08 | 0.0 | 0.0 |
| CD24−CD44+CD49F+ | ND | 26.00 | 29.78 | 25.36 | 0.0 | 0.0 |
| CD24−CD44+CD49F- | ND | 7.50 | 34.08 | 19.06 | 0.0 | 0.0 |
| CD24+CD44+CD49F+EpCAM+ | ND | 1.25 | 2.36 | 11.68 | 0.0 | 0.0 |
| CD24+CD44+CD49f+ EpCAM- | ND | 4.42 | 5.12 | 11.74 | 0.0 | 0.0 |
| CD24−CD44+CD49F+EpCAM+ | ND | 5.94 | 29.78 | 25.36 | 0.0 | 0.0 |
| CD24−CD44+CD49F+EpCAM- | ND | 20.00 | 32 | 15.36 | 0.0 | 0.0 |
| CD24−CD44+CD49f+EpCAM+ALDH1+ | ND | 6.0 | 30.08 | 25.46 | 0.0 | 0.0 |
| CD24−CD44+CD49f+EpCAM-ALDH1+ | ND | 3.0 | 2.08 | 0.0 | 0.0 | 0.0 |
| CD24+CD44+CD49f+ALDH+CXCR4+ | ND | 1.2 | 2.84 | 12.0 | 0.0 | 0.0 |
| CD24+CD44+CD49f+ALDH-CXCR4+ | ND | 3.3 | 2.24 | 35.8 | 0.0 | 0.0 |
| CD24−CD44+CD49f+ALDH+CXCR4+ | ND | 1.3 | 30.00 | 25.20 | 0.0 | 0.0 |
| CD24−CD44+CD49f+ALDH-CXCR4+ | ND | 8.0 | 42.65 | 5.20 | 0.0 | 0.0 |
| CD24−CD44+CD49f+ALDH+CXCR4+CD133+ | ND | 1.3 | 30.00 | 25.20 | 0.0 | 0.0 |
| CD24+CD44+CD49f+ALDH+CXCR4+NRF1+ | ND | 1.2 | 2.76 | 56.60 | 0.0 | 0.0 |
| CD24−CD44+CD49f+ALDH+CXCR4+NRF1+ | ND | 1.3 | 30.59 | 25.10 | 0.0 | 0.0 |
| CD24−CD44+CD49f+ALDH-CXCR4-NRF1+ | ND | 0.0 | 0.0 | 15.10 | 0.0 | 0.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, J.K.; Felty, Q.; Poppiti, R.; Jackson, R.M.; Roy, D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells 2018, 7, 234. https://doi.org/10.3390/cells7120234
Das JK, Felty Q, Poppiti R, Jackson RM, Roy D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells. 2018; 7(12):234. https://doi.org/10.3390/cells7120234
Chicago/Turabian StyleDas, Jayanta K., Quentin Felty, Robert Poppiti, Robert M. Jackson, and Deodutta Roy. 2018. "Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis" Cells 7, no. 12: 234. https://doi.org/10.3390/cells7120234
APA StyleDas, J. K., Felty, Q., Poppiti, R., Jackson, R. M., & Roy, D. (2018). Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells, 7(12), 234. https://doi.org/10.3390/cells7120234