Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease

Abstract
1. Introduction
2. AQP4 and Its Role during Development
3. AQP4 and Its Role in the Adult Brain
Insights from AQP4 Deficient Mice
4. AQP4 during Disease
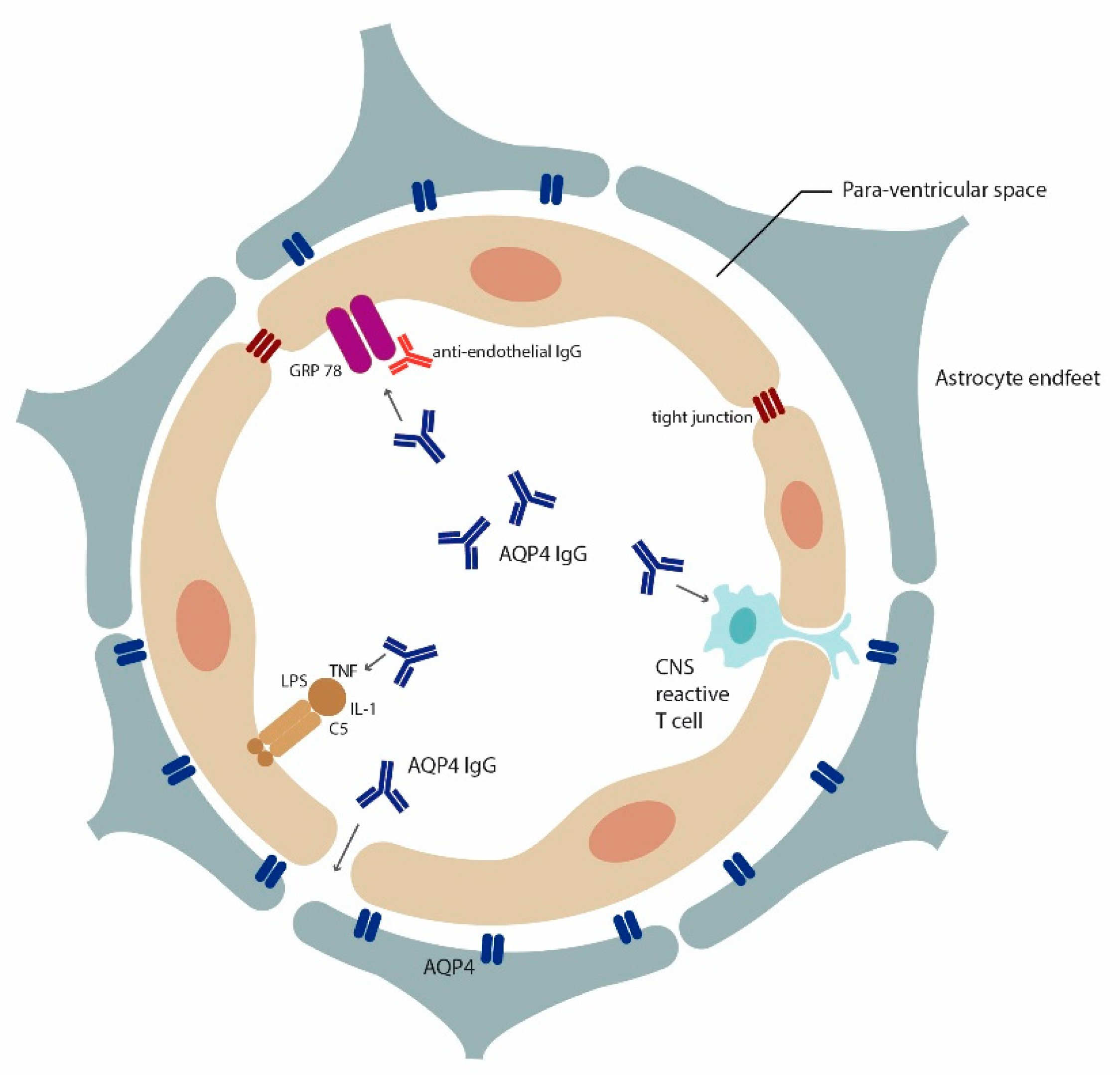
4.1. Role of AQP4 in Inflammation
Autoantibodies to Astrocytic AQP4 in NMOSD and Its Disease Spectrum
4.2. Role of AQP4 in Controlling Brain-Water Balance
AQP4 and Hydrocephalus
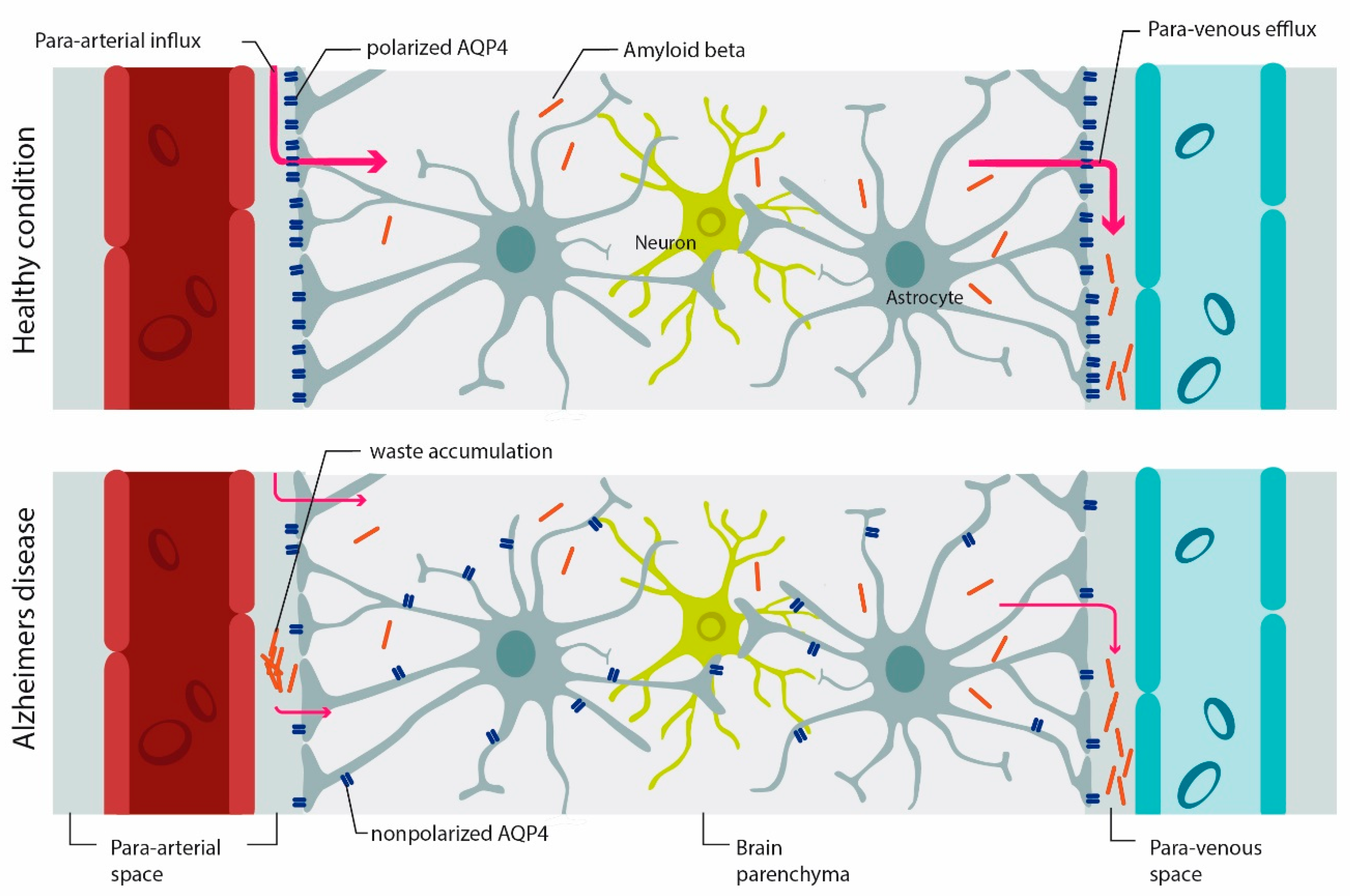
4.3. Role of AQP4 in Glymphatic Clearance, Synaptic Plasticity and Memory
AQP4 and Alzheimer Diseases
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Badaut, J.; Brunet, J.F.; Regli, L. Aquaporins in the brain: From aqueduct to “multi-duct”. Metab. Brain Dis. 2007, 22, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Yasumura, T.; Hudson, C.S.; Agre, P.; Nielsen, S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl. Acad. Sci. USA 1998, 95, 11981–11986. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Ma, T.; Skach, W.; Matthay, M.A.; Verkman, A.S. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 1994, 269, 5497–5500. [Google Scholar] [PubMed]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Murlidharan, G.; Crowther, A.; Reardon, R.A.; Song, J.; Asokan, A. Glymphatic fluid transport controls paravascular clearance of AAV vectors from the brain. JCI Insight 2016, 1, e88034. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Binder, D.K.; Bloch, O.; Auguste, K.; Papadopoulos, M.C. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim. Biophys. Acta 2006, 1758, 1085–1093. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef]
- Zhao, Z.A.; Li, P.; Ye, S.Y.; Ning, Y.L.; Wang, H.; Peng, Y.; Yang, N.; Zhao, Y.; Zhang, Z.H.; Chen, J.F.; et al. Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A2A receptor inactivation. Sci. Rep. 2017, 7, 2254. [Google Scholar] [CrossRef]
- Aoki, K.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003, 106, 121–124. [Google Scholar] [CrossRef]
- Hubbard, J.A.; Szu, J.I.; Binder, D.K. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res. Bull. 2018, 136, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [PubMed]
- Aprea, J.; Calegari, F. Long non-coding RNAs in corticogenesis: Deciphering the non-coding code of the brain. EMBO J. 2015, 34, 2865–2884. [Google Scholar] [CrossRef] [PubMed]
- Fallier-Becker, P.; Vollmer, J.P.; Bauer, H.C.; Noell, S.; Wolburg, H.; Mack, A.F. Onset of aquaporin-4 expression in the developing mouse brain. Int. J. Dev. Neurosci. 2014, 36, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Papadopoulos, M.C.; Liu, J.; Li, L.; Zhang, D.; Zhang, H.; Verkman, A.S.; Ma, T. Sporadic obstructive hydrocephalus in Aqp4 null mice. J. Neurosci. Res. 2009, 87, 1150–1155. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef]
- Walz, T.; Fujiyoshi, Y.; Engel, A. The AQP structure and functional implications. Handb. Exp. Pharmacol. 2009, 31–56. [Google Scholar]
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.; Robbins, R.A.; Miercke, L.J.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442. [Google Scholar] [CrossRef]
- Lu, M.; Lee, M.D.; Smith, B.L.; Jung, J.S.; Agre, P.; Verdijk, M.A.; Merkx, G.; Rijss, J.P.; Deen, P.M. The human AQP4 gene: Definition of the locus encoding two water channel polypeptides in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 10908–10912. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Moritz, T.J.; Ratelade, J.; Verkman, A.S. Super-resolution imaging of aquaporin-4 orthogonal arrays of particles in cell membranes. J. Cell Sci. 2012, 125, 4405–4412. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.S.; Gorelick-Feldman, D.A.; Davidson, K.G.; Yasumura, T.; Neely, J.D.; Agre, P.; Rash, J.E. Aquaporin-4 square array assembly: Opposing actions of M1 and M23 isoforms. Proc. Natl. Acad. Sci. USA 2003, 100, 13609–13614. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.M.; Bennett, J.L.; Verkman, A.S. Live cell analysis of aquaporin-4 m1/m23 interactions and regulated orthogonal array assembly in glial cells. J. Biol. Chem. 2009, 284, 35850–35860. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; van Hoek, A.N.; Verkman, A.S. Very high single channel water permeability of aquaporin-4 in baculovirus-infected insect cells and liposomes reconstituted with purified aquaporin-4. Biochemistry 1997, 36, 7625–7632. [Google Scholar] [CrossRef] [PubMed]
- Hiroaki, Y.; Tani, K.; Kamegawa, A.; Gyobu, N.; Nishikawa, K.; Suzuki, H.; Walz, T.; Sasaki, S.; Mitsuoka, K.; Kimura, K.; et al. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2006, 355, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, C.; Bouley, R.; Huang, Y.; Fang, P.; Pastor-Soler, N.; Brown, D.; Van Hoek, A.N. Membrane organization and function of M1 and M23 isoforms of aquaporin-4 in epithelial cells. Am. J. Physiol. Renal Physiol. 2004, 287, F501–F511. [Google Scholar] [CrossRef]
- Verkman, A.S.; Ratelade, J.; Rossi, A.; Zhang, H.; Tradtrantip, L. Aquaporin-4: Orthogonal array assembly, CNS functions, and role in neuromyelitis optica. Acta Pharmacol. Sin. 2011, 32, 702–710. [Google Scholar] [CrossRef]
- Mader, S.; Lutterotti, A.; Di Pauli, F.; Kuenz, B.; Schanda, K.; Aboul-Enein, F.; Khalil, M.; Storch, M.K.; Jarius, S.; Kristoferitsch, W.; et al. Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS ONE 2010, 5, e10455. [Google Scholar] [CrossRef]
- Nicchia, G.P.; Mastrototaro, M.; Rossi, A.; Pisani, F.; Tortorella, C.; Ruggieri, M.; Lia, A.; Trojano, M.; Frigeri, A.; Svelto, M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 2009, 57, 1363–1373. [Google Scholar] [CrossRef]
- Crane, J.M.; Lam, C.; Rossi, A.; Gupta, T.; Bennett, J.L.; Verkman, A.S. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J. Biol. Chem. 2011, 286, 16516–16524. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Abe, Y.; Iwanari, H.; Suzuki, Y.; Kikuchi, T.; Ito, T.; Kato, J.; Kusano-Arai, O.; Takahashi, T.; Nishiyama, S.; et al. Establishment of monoclonal antibodies against the extracellular domain that block binding of NMO-IgG to AQP4. J. Neuroimmunol. 2013, 260, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Krishna, S.; Bell, B.A. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatry 2002, 72, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Solenov, E.; Watanabe, H.; Manley, G.T.; Verkman, A.S. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol. Cell Physiol. 2004, 286, C426–C432. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, O.; Laake, P.; Klungland, A.; Thoren, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [PubMed]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 2011, 31, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Chen, Z.G.; Dang, H.; Ding, J.H.; Fan, Y.; Hu, G. Glia protein aquaporin-4 regulates aversive motivation of spatial memory in Morris water maze. CNS Neurosci. Ther. 2013, 19, 937–944. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef]
- Saadoun, S.; Tait, M.J.; Reza, A.; Davies, D.C.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience 2009, 161, 764–772. [Google Scholar] [CrossRef]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Manley, G.T.; Krishna, S.; Verkman, A.S. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004, 18, 1291–1293. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Probst, C.; Borowski, K.; Franciotta, D.; Wildemann, B.; Stoecker, W.; Wandinger, K.P. Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J. Neurol. Sci. 2010, 291, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; Wingerchuk, D.M.; Vukusic, S.; Linbo, L.; Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann. Neurol. 2006, 59, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Palace, J.; Leite, M.I.; Nairne, A.; Vincent, A. Interferon Beta treatment in neuromyelitis optica: Increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010, 67, 1016–1017. [Google Scholar] [CrossRef]
- Waters, P.; Reindl, M.; Saiz, A.; Schanda, K.; Tuller, F.; Kral, V.; Nytrova, P.; Sobek, O.; Nielsen, H.H.; Barington, T.; et al. Multicentre comparison of a diagnostic assay: Aquaporin-4 antibodies in neuromyelitis optica. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1005–1015. [Google Scholar] [CrossRef]
- Waters, P.J.; McKeon, A.; Leite, M.I.; Rajasekharan, S.; Lennon, V.A.; Villalobos, A.; Palace, J.; Mandrekar, J.N.; Vincent, A.; Bar-Or, A.; et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology 2012, 78, 665–671; discussion 669. [Google Scholar] [CrossRef]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: A study on antibody titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Gredler, V.; Schanda, K.; Rostasy, K.; Dujmovic, I.; Pfaller, K.; Lutterotti, A.; Jarius, S.; Di Pauli, F.; Kuenz, B.; et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflamm. 2011, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Kitley, J.; Woodhall, M.; Leite, M.I.; Palace, J.; Vincent, A.; Waters, P. Aquaporin-4 antibody isoform binding specificities do not explain clinical variations in NMO. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e121. [Google Scholar] [CrossRef] [PubMed]
- Di Pauli, F.; Mader, S.; Rostasy, K.; Schanda, K.; Bajer-Kornek, B.; Ehling, R.; Deisenhammer, F.; Reindl, M.; Berger, T. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin. Immunol. 2011, 138, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Phuan, P.W.; Asavapanumas, N.; Tradtrantip, L. Biology of AQP4 and anti-AQP4 antibody: Therapeutic implications for NMO. Brain Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Fryer, J.P.; Lennon, V.A.; Pittock, S.J.; Jenkins, S.M.; Fallier-Becker, P.; Clardy, S.L.; Horta, E.; Jedynak, E.A.; Lucchinetti, C.F.; Shuster, E.A.; et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e11. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Takai, Y.; Kusano-Arai, O.; Ramadhanti, J.; Iwanari, H.; Miyauchi, T.; Sakihama, T.; Han, J.Y.; Aoki, M.; Hamakubo, T.; et al. The binding property of a monoclonal antibody against the extracellular domains of aquaporin-4 directs aquaporin-4 toward endocytosis. Biochem. Biophys. Rep. 2016, 7, 77–83. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef]
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009, 66, 630–643. [Google Scholar] [CrossRef]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef]
- Pohl, M.; Kawakami, N.; Kitic, M.; Bauer, J.; Martins, R.; Fischer, M.T.; Machado-Santos, J.; Mader, S.; Ellwart, J.W.; Misu, T.; et al. T cell-activation in neuromyelitis optica lesions plays a role in their formation. Acta Neuropathol. Commun. 2013, 1, 85. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Fischer, M.T.; Mader, S.; Schanda, K.; Kitic, M.; Sharma, R.; Wimmer, I.; Misu, T.; Fujihara, K.; Reindl, M.; et al. Pathogenic T cell responses against aquaporin 4. Acta Neuropathol. 2011, 122, 21–34. [Google Scholar] [CrossRef]
- Jones, M.V.; Huang, H.; Calabresi, P.A.; Levy, M. Pathogenic aquaporin-4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol. Commun. 2015, 3, 28. [Google Scholar] [CrossRef]
- Shimizu, F.; Schaller, K.L.; Owens, G.P.; Cotleur, A.C.; Kellner, D.; Takeshita, Y.; Obermeier, B.; Kryzer, T.J.; Sano, Y.; Kanda, T.; et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med. 2017, 9, eaai9111. [Google Scholar] [CrossRef]
- Hillebrand, S.; Schanda, K.; Nigritinou, M.; Tsymala, I.; Bohm, D.; Peschl, P.; Takai, Y.; Fujihara, K.; Nakashima, I.; Misu, T.; et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol. 2018, 1–19. [Google Scholar]
- Takeshita, Y.; Obermeier, B.; Cotleur, A.C.; Spampinato, S.F.; Shimizu, F.; Yamamoto, E.; Sano, Y.; Kryzer, T.J.; Lennon, V.A.; Kanda, T.; et al. Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e311. [Google Scholar] [CrossRef]
- Yick, L.W.; Ma, O.K.; Ng, R.C.; Kwan, J.S.; Chan, K.H. Aquaporin-4 Autoantibodies from Neuromyelitis Optica Spectrum Disorder Patients Induce Complement-Independent Immunopathologies in Mice. Front. Immunol. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Nishiyama, S.; Misu, T.; Nuriya, M.; Takano, R.; Takahashi, T.; Nakashima, I.; Yasui, M.; Itoyama, Y.; Aoki, M.; Fujihara, K. Complement-dependent and -independent aquaporin 4-antibody-mediated cytotoxicity in human astrocytes: Pathogenetic implications in neuromyelitis optica. Biochem. Biophys. Rep. 2016, 7, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125, 1450–1461. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Treatment of neuromyelitis optica: State-of-the-art and emerging therapies. Nat. Rev. Neurol. 2014, 10, 493–506. [Google Scholar] [CrossRef]
- Bergman, I.; Basse, P.H.; Barmada, M.A.; Griffin, J.A.; Cheung, N.K. Comparison of in vitro antibody-targeted cytotoxicity using mouse, rat and human effectors. Cancer Immunol. Immunother. 2000, 49, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Nytrova, P.; Potlukova, E.; Kemlink, D.; Woodhall, M.; Horakova, D.; Waters, P.; Havrdova, E.; Zivorova, D.; Vincent, A.; Trendelenburg, M. Complement activation in patients with neuromyelitis optica. J. Neuroimmunol. 2014, 274, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Hoftberger, R.; Fujihara, K.; Wimmer, I.; Takai, Y.; Nishiyama, S.; Nakashima, I.; Konno, H.; Bradl, M.; Garzuly, F.; et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013, 125, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Ratelade, J.; Asavapanumas, N.; Ritchie, A.M.; Wemlinger, S.; Bennett, J.L.; Verkman, A.S. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013, 126, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216. [Google Scholar] [CrossRef]
- Cree, B.A.; Lamb, S.; Morgan, K.; Chen, A.; Waubant, E.; Genain, C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005, 64, 1270–1272. [Google Scholar] [CrossRef]
- Gredler, V.; Mader, S.; Schanda, K.; Hegen, H.; Di Pauli, F.; Kuenz, B.; Deisenhammer, F.; Berger, T.; Reindl, M.; Lutterotti, A. Clinical and immunological follow-up of B-cell depleting therapy in CNS demyelinating diseases. J. Neurol. Sci. 2013, 328, 77–82. [Google Scholar] [CrossRef]
- Mealy, M.A.; Kim, S.H.; Schmidt, F.; Lopez, R.; Jimenez Arango, J.A.; Paul, F.; Wingerchuk, D.M.; Greenberg, B.M.; Kim, H.J.; Levy, M. Aquaporin-4 serostatus does not predict response to immunotherapy in neuromyelitis optica spectrum disorders. Mult. Scler. 2018, 24, 1737–1742. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Souliotis, V.L.; Fragiadaki, K.G.; Moutsopoulos, H.M.; Boletis, J.N.; Theofilopoulos, A.N. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin. Immunol. 2007, 123, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.A.; Athanazio, D.A.; Lima, I.; Oliveira e Silva, N.; Andrade, J.C.; Jesus, R.N.; Barbosa, L.M.; Reis, M.G.; Santiago, M.B. NK and NKT cell dynamics after rituximab therapy for systemic lupus erythematosus and rheumatoid arthritis. Rheumatol. Int. 2009, 29, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012, 71, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Soltys, J.N.; Meyer, S.A.; Schumann, H.; Gibson, E.A.; Restrepo, D.; Bennett, J.L. Determining the Spatial Relationship of Membrane-Bound Aquaporin-4 Autoantibodies by STED Nanoscopy. Biophys. J. 2017, 112, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Enno, T.L.; Del Bigio, M.R. Aquaporin 4 changes in rat brain with severe hydrocephalus. Eur. J. Neurosci. 2006, 23, 2929–2936. [Google Scholar] [CrossRef] [PubMed]
- Skjolding, A.D.; Rowland, I.J.; Sogaard, L.V.; Praetorius, J.; Penkowa, M.; Juhler, M. Hydrocephalus induces dynamic spatiotemporal regulation of aquaporin-4 expression in the rat brain. Cereb. Fluid Res. 2010, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Tourdias, T.; Dragonu, I.; Fushimi, Y.; Deloire, M.S.; Boiziau, C.; Brochet, B.; Moonen, C.; Petry, K.G.; Dousset, V. Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: A combined MRI-histological study. Neuroimage 2009, 47, 659–666. [Google Scholar] [CrossRef]
- Bloch, O.; Auguste, K.I.; Manley, G.T.; Verkman, A.S. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J. Cereb. Blood Flow Metab. 2006, 26, 1527–1537. [Google Scholar] [CrossRef]
- Castaneyra-Ruiz, L.; Gonzalez-Marrero, I.; Gonzalez-Toledo, J.M.; Castaneyra-Ruiz, A.; de Paz-Carmona, H.; Castaneyra-Perdomo, A.; Carmona-Calero, E.M. Aquaporin-4 expression in the cerebrospinal fluid in congenital human hydrocephalus. Fluids Barriers CNS 2013, 10, 18. [Google Scholar] [CrossRef]
- Clardy, S.L.; Lucchinetti, C.F.; Krecke, K.N.; Lennon, V.A.; O’Toole, O.; Weinshenker, B.G.; Boyd, C.D.; Krieger, S.; McGraw, C.; Guo, Y.; et al. Hydrocephalus in neuromyelitis optica. Neurology 2014, 82, 1841–1843. [Google Scholar] [CrossRef]
- Magana, S.M.; Matiello, M.; Pittock, S.J.; McKeon, A.; Lennon, V.A.; Rabinstein, A.A.; Shuster, E.; Kantarci, O.H.; Lucchinetti, C.F.; Weinshenker, B.G. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 2009, 72, 712–717. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Shokri-Kojori, E.; Wang, G.J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Smith, S.R.; Mazzucchelli, G.N.; Villemagne, V.L.; Brown, B.M.; Porter, T.; Weinborn, M.; Bucks, R.S.; Milicic, L.; Sohrabi, H.R.; Taddei, K.; et al. Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl. Psychiatry 2018, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Verkman, A.S. The “glymphatic” mechanism for solute clearance in Alzheimer’s disease: Game changer or unproven speculation? FASEB J. 2018, 32, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, M.X.; Luo, Y.; Chen, T.; Liu, J.; Fang, P.; Jiang, B.; Hu, Z.L.; Jin, Y.; Chen, J.G.; et al. Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology 2013, 75, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.N.; Sun, X.L.; Gao, L.; Fan, Y.; Ding, J.H.; Hu, G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol. Cell. Neurosci. 2007, 34, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Potential utility of aquaporin modulators for therapy of brain disorders. Prog. Brain Res. 2008, 170, 589–601. [Google Scholar]
- Pirici, I.; Balsanu, T.A.; Bogdan, C.; Margaritescu, C.; Divan, T.; Vitalie, V.; Mogoanta, L.; Pirici, D.; Carare, R.O.; Muresanu, D.F. Inhibition of Aquaporin-4 Improves the Outcome of Ischaemic Stroke and Modulates Brain Paravascular Drainage Pathways. Int. J. Mol. Sci. 2017, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Tradtrantip, L.; Felix, C.M.; Spirig, R.; Morelli, A.B.; Verkman, A.S. Recombinant IgG1 Fc hexamers block cytotoxicity and pathological changes in experimental in vitro and rat models of neuromyelitis optica. Neuropharmacology 2018, 133, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Foglio, E.; Rodella, L.F. Aquaporins and neurodegenerative diseases. Curr. Neuropharmacol. 2010, 8, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Matsumoto, S.; Moriwaki, Y.; Okuda, T.; Ohara, S.; Yamanaka, K.; Abe, Y.; Yasui, M.; Misawa, H. Dissociation of blood-brain barrier disruption and disease manifestation in an aquaporin-4-deficient mouse model of amyotrophic lateral sclerosis. Neurosci. Res. 2018, 133, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Prydz, A.; Stahl, K.; Puchades, M.; Davarpaneh, N.; Nadeem, M.; Ottersen, O.P.; Gundersen, V.; Amiry-Moghaddam, M. Subcellular expression of aquaporin-4 in substantia nigra of normal and MPTP-treated mice. Neuroscience 2017, 359, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Tsunoda, A.; Tada, M.; Nishizawa, M.; Ugawa, Y.; Kakita, A. Expression of Aquaporin 1 and Aquaporin 4 in the Temporal Neocortex of Patients with Parkinson’s Disease. Brain Pathol. 2017, 27, 160–168. [Google Scholar] [CrossRef]
- Fan, Y.; Kong, H.; Shi, X.; Sun, X.; Ding, J.; Wu, J.; Hu, G. Hypersensitivity of aquaporin 4-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine and astrocytic modulation. Neurobiol. Aging 2008, 29, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.; Hsu, Y.; Schneller, B.; Hobbs, J.G.; Mehta, A.I.; Linninger, A. Hydrocephalus: The role of cerebral aquaporin-4 channels and computational modeling considerations of cerebrospinal fluid. Neurosurg. Focus 2016, 41, E8. [Google Scholar] [CrossRef] [PubMed]
- Tome-Garcia, J.; Erfani, P.; Nudelman, G.; Tsankov, A.M.; Katsyv, I.; Tejero, R.; Bin, Z.; Walsh, M.; Friedel, R.H.; Zaslavsky, E.; et al. Analysis of chromatin accessibility uncovers TEAD1 as a regulator of migration in human glioblastoma. Nat. Commun. 2018, 9, 4020. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, R.; Schiera, G.; Di Liegro, C.M.; Fricano, A.; Iacopino, D.G.; Di Liegro, I. Aquaporins and Brain Tumors. Int. J. Mol. Sci. 2016, 17, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.; Byne, W.; Roussos, P.; Tan, W.; Siever, L.; Haroutunian, V. Astrocyte and glutamate markers in the superficial, deep, and white matter layers of the anterior cingulate gyrus in schizophrenia. Neuropsychopharmacology 2011, 36, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Hughes, J.; Stockmeier, C.A.; Javier Miguel-Hidalgo, J.; Maciag, D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 2013, 73, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Eid, T.; Mane, S.; Kim, J.H.; Spencer, D.D.; Ottersen, O.P.; de Lanerolle, N.C. Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 2004, 108, 493–502. [Google Scholar] [CrossRef]
- Binder, D.K.; Oshio, K.; Ma, T.; Verkman, A.S.; Manley, G.T. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 2004, 15, 259–262. [Google Scholar] [CrossRef]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 2014, 5, 3. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Mechanism | AQP4 Related Pathology | Rodent Models | (Potential) Therapeutic |
|---|---|---|---|---|
| NMOSD | -AQP4-IgG dependent tissue astrocytophathy: -CDC -ADCC -other mechanisms | -Presence of AQP4-IgG in serum [44] -Loss of AQP4 in NMOSD lesions [105] -Massive demyelination (brain, optic nerve, spinal cord) [70,75] | 1. Injection of anti-AQP4-IgG into the brain [73] 2. Intravenous injection of AQP4-IgG following BBB disruption through T cell- mediated EAE [59,60], bacterial proteins [67] or anti-endothelial antibody [64]. 3. High affinity circulating rodent AQP4-IgG enter the CNS without BBB impairment [65] | -Decoy antibody lacking FcR or complement binding [84] -Recombinant IgG1 Fc hexamers that block cytotoxicity and pathological changes [106] |
| Alzheimer’s Disease | Loss of AQP4 polarization with impaired clearance of interstitial solutes and increased aggregation (beta- amyloid) | Mislocalization of AQP4 [13] | AQP4 gene knockout of beta-amyloid precursor protein /presenilin 1 (APP/PS1) transgenic mice [107] | AQP4 receptor agonists [107] |
| ALS | Loss of AQP4 polarization and altered AQP4 expression is contributing to motor neuron degeneration [107] and BBB impairment [108] | AQP4 overexpression in astrocytes [109] | Superoxide dismutase 1 (SOD1) G93A transgenic mice (mouse model of ALS) [109] | Targeting AQP4 as potential treatment to restore BBB in ALS [108] |
| Parkinson’s Disease (PD) | AQP4 dysfunction contributing to synuclein deposition and water accumulation in the substantia nigra [110] | Enriched AQP4-positive astrocytes in the neocortex [111] | AQP4 deficient mice treated with MPTP [112] | N/A |
| Ischemic Stroke | AQP4 enhances edema formation or diminishes resolution | Enhanced expression of AQP4 at site of infarction [10]. | Brain edema caused by acute water intoxication using AQP4 knock out mice [41] | AQP4 inhibitors during edema formation |
| Traumatic brain injury (TBI) | AQP4 is altering water homeostasis and AQP4 may be associated with neuroinflammation (through astrocyte and microglia activation) | Increased expression of AQP4 and loss of AQP4 polarity [9] | TBI mouse model [9] | AQP4 inhibitors may be beneficial [103] |
| Hydrocephalus | Control of water homeostasis | -Increased AQP4 in CSF of congenital communicating hydrocephalus [90], -Hydrocephalus has been reported in AQP4-IgG positive NMOSD [91] | Rat kaolin model [86] | Increasing AQP4 to support CSF clearance at a later disease stage or decreasing AQP4 in areas of CSF production particularly at disease onset [113]. |
| Glioma | -AQP4 is contributing to increased tumor cell migration possibly through increasing water permeability [114]. -Involvement of AQP4 in tumor edema | Expression of AQP4 in human glioblastoma [115] | N/A | Use of AQP inhibitors to reduce tumor growth [18] |
| Schizophrenia | Reduced AQP4 is contributing to neurovascular dysfunction and BBB hyperpermeability | Astroglial loss and reduced AQP4 expression in the deep layers of the anterior cingulated gyrus [116] | N/A | N/A |
| Major depressive disorder (MDD) | AQP4 is contributing to poor water balance | Reduced coverage of blood vessels by AQP4 positive astrocytic endfeet in the orbitofrontal cortex [117] | N/A | N/A |
| Epilepsy | Impairment of K+ homeostasis | AQP4 expression is increased in samples from atrophic hippocampus from epileptic patients [118] | AQP4 deficient mice [119] | AQP4 modulators to increase seizure thresholds [103] |
| Autism | Abnormal glial-neuronal communication in brains of subjects with autism | Decreased AQP4 expression in cerebellum of post mortem tissue [120] | N/A | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. https://doi.org/10.3390/cells8020090
Mader S, Brimberg L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells. 2019; 8(2):90. https://doi.org/10.3390/cells8020090
Chicago/Turabian StyleMader, Simone, and Lior Brimberg. 2019. "Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease" Cells 8, no. 2: 90. https://doi.org/10.3390/cells8020090
APA StyleMader, S., & Brimberg, L. (2019). Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells, 8(2), 90. https://doi.org/10.3390/cells8020090