Mitochondrial DNA Integrity: Role in Health and Disease


Abstract
1. Introduction
2. Mitochondrial DNA: Structure
3. Mitochondrial DNA: Transcription and Replication
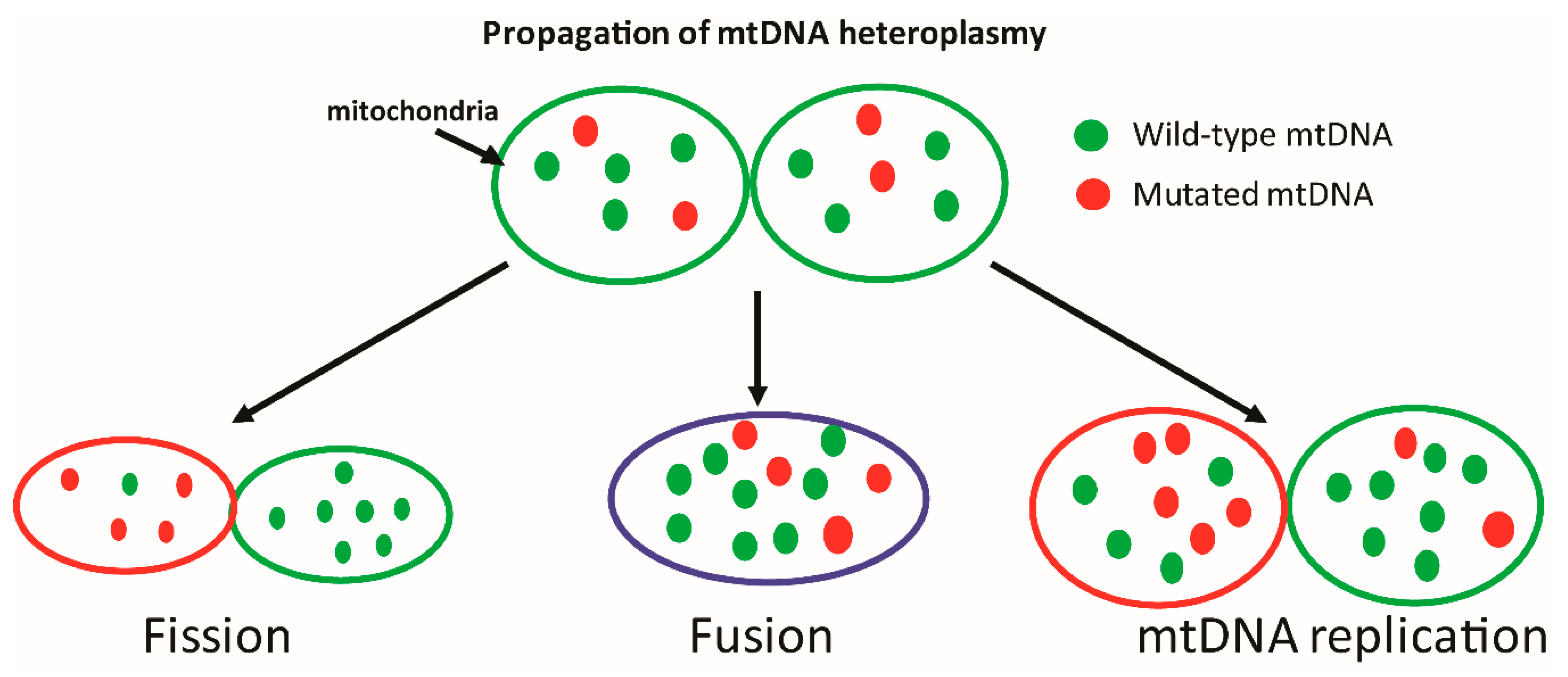
4. Induction of Mitochondrial DNA Damage
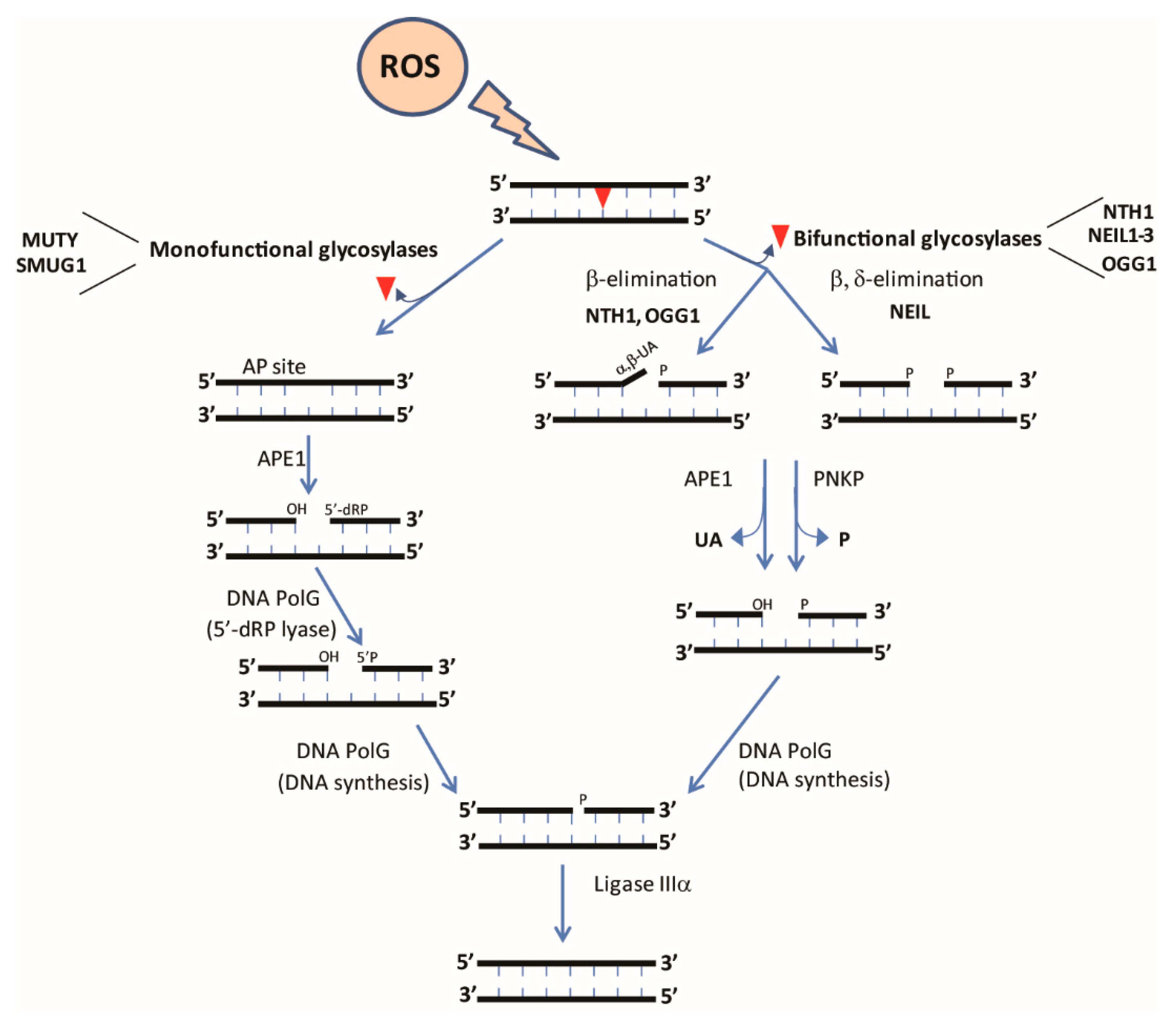
5. Base-Excision Repair of Mitochondrial DNA
6. Impact of Oxidative Damage on Mitochondrial Replication and Transcription
7. Pathologies Associated with Mitochondrial Repair Defects
8. Cancer
9. Neurodegenerative Diseases: Parkinson’s and Alzheimer’s
10. Metabolic Disease
11. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Ohkubo, K.; Yamano, A.; Nagashima, M.; Mori, Y.; Anzai, K.; Akehi, Y.; Nomiyama, R.; Asano, T.; Urae, A.; Ono, J. Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and diabetes: Prevalence and clinical phenotypes in Japan. Clin. Chem. 2001, 47, 1641–1648. [Google Scholar] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Huoponen, K. Leber hereditary optic neuropathy: Clinical and molecular genetic findings. Neurogenetics 2001, 3, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Nakano, S.; Imaizumi, N.; Kitazawa, M.; Nishizawa, M.; Kigoshi, T.; Uchida, K. Mitochondrial DNA mutations are associated with both decreased insulin secretion and advanced microvascular complications in Japanese diabetic subjects. J. Diabetes Its Complicat. 1999, 13, 277–283. [Google Scholar] [CrossRef]
- Goldring, E.S.; Grossman, L.I.; Krupnick, D.; Cryer, D.R.; Marmur, J. The petite mutation in yeast. Loss of mitochondrial deoxyribonucleic acid during induction of petites with ethidium bromide. J. Mol. Biol. 1970, 52, 323–335. [Google Scholar] [CrossRef]
- Desjardins, P.; Frost, E.; Morais, R. Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. Mol. Cell. Biol. 1985, 5, 1163–1169. [Google Scholar] [CrossRef]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Jones, E.; Ramos, M.; Innis-Whitehouse, W.; Gilkerson, R. The little big genome: The organization of mitochondrial DNA. Front. Biosci. 2017, 22, 710–721. [Google Scholar]
- Hillar, M.; Rangayya, V.; Jafar, B.B.; Chambers, D.; Vitzu, M.; Wyborny, L.E. Membrane-bound mitochondrial DNA: Isolation, transcription and protein. Arch. Int. Physiol. Biochim. 1979, 87, 29–49. [Google Scholar]
- Chen, X.J.; Butow, R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Moraes, C.T.; Srivastava, S.; Kirkinezos, I.; Oca-Cossio, J.; van Waveren, C.; Woischnick, M.; Diaz, F. Mitochondrial DNA structure and function. Int. Rev. Neurobiol. 2002, 53, 3–23. [Google Scholar]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, H.; Vinograd, J. Replication of circular DNA in eukaryotic cells. Annu. Rev. Biochem. 1974, 43, 695–719. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef]
- Stoneking, M. Hypervariable sites in the mtDNA control region are mutational hotspots. Am. J. Hum. Genet. 2000, 67, 1029–1032. [Google Scholar] [CrossRef]
- Chocron, E.S.; Munkacsy, E.; Pickering, A.M. Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 9, 285–297. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Arnberg, A.; van Bruggen, E.F.; Borst, P. The presence of DNA molecules with a displacement loop in standard mitochondrial DNA preparations. Biochim. Biophys. Acta 1971, 246, 353–357. [Google Scholar] [CrossRef]
- Kasamatsu, H.; Robberson, D.L.; Vinograd, J. A novel closed-circular mitochondrial DNA with properties of a replicating intermediate. Proc. Natl. Acad. Sci. USA 1971, 68, 2252–2257. [Google Scholar] [CrossRef]
- Ingman, M.; Kaessmann, H.; Paabo, S.; Gyllensten, U. Mitochondrial genome variation and the origin of modern humans. Nature 2000, 408, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.T.; Sun, Y.; Bu, L.X.; Jia, M.Y. Gene mutations in the D-loop region of mitochondrial DNA in oral squamous cell carcinoma. Mol. Med. Rep. 2015, 11, 4496–4500. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Singh, A.; Sharma, C.; Jain, S.K.; Singh, N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005, 5, 34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miyazono, F.; Schneider, P.M.; Metzger, R.; Warnecke-Eberz, U.; Baldus, S.E.; Dienes, H.P.; Aikou, T.; Hoelscher, A.H. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene 2002, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.Y.; Lin, J.C.; Lee, L.M.; Wu, C.W.; Tseng, L.M.; Yin, P.H.; Chi, C.W.; Lee, H.C. Tandem duplication/triplication correlated with poly-cytosine stretch variation in human mitochondrial DNA D-loop region. Mutagenesis 2008, 23, 137–142. [Google Scholar] [CrossRef]
- Guo, X.G.; Guo, Q.N. Mutations in the mitochondrial DNA D-Loop region occur frequently in human osteosarcoma. Cancer Lett. 2006, 239, 151–155. [Google Scholar] [CrossRef]
- Wang, Y.; Michikawa, Y.; Mallidis, C.; Bai, Y.; Woodhouse, L.; Yarasheski, K.E.; Miller, C.A.; Askanas, V.; Engel, W.K.; Bhasin, S.; et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4022–4027. [Google Scholar] [CrossRef]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial transcription in mammalian cells. Front. Biosci. 2017, 22, 835–853. [Google Scholar]
- Montoya, J.; Christianson, T.; Levens, D.; Rabinowitz, M.; Attardi, G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 7195–7199. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef] [PubMed]
- Asin-Cayuela, J.; Gustafsson, C.M. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci. 2007, 32, 111–117. [Google Scholar] [CrossRef]
- Cantatore, P.; Attardi, G. Mapping of nascent light and heavy strand transcripts on the physical map of HeLa cell mitochondrial DNA. Nucleic Acids Res. 1980, 8, 2605–2625. [Google Scholar] [CrossRef]
- Sanchez, M.I.; Mercer, T.R.; Davies, S.M.; Shearwood, A.M.; Nygard, K.K.; Richman, T.R.; Mattick, J.S.; Rackham, O.; Filipovska, A. RNA processing in human mitochondria. Cell Cycle 2011, 10, 2904–2916. [Google Scholar] [CrossRef]
- Martin, M.; Cho, J.; Cesare, A.J.; Griffith, J.D.; Attardi, G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 2005, 123, 1227–1240. [Google Scholar] [CrossRef]
- Chang, D.D.; Clayton, D.A. Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell 1984, 36, 635–643. [Google Scholar] [CrossRef]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural basis of mitochondrial transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef]
- Guja, K.E.; Garcia-Diaz, M. Hitting the brakes: Termination of mitochondrial transcription. Biochim. Biophys. Acta 2012, 1819, 939–947. [Google Scholar] [CrossRef]
- Adan, C.; Matsushima, Y.; Hernandez-Sierra, R.; Marco-Ferreres, R.; Fernandez-Moreno, M.A.; Gonzalez-Vioque, E.; Calleja, M.; Aragon, J.J.; Kaguni, L.S.; Garesse, R. Mitochondrial transcription factor B2 is essential for metabolic function in Drosophila melanogaster development. J. Biol. Chem. 2008, 283, 12333–12342. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.L.; Shadel, G.S. Accessorizing the human mitochondrial transcription machinery. Trends Biochem. Sci. 2013, 38, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Dillon, L.M.; Moraes, C.T. Mitochondrial DNA transcription regulation and nucleoid organization. J. Inherit. Metab. Dis. 2011, 34, 941–951. [Google Scholar] [CrossRef]
- Wang, Z.; Cotney, J.; Shadel, G.S. Human mitochondrial ribosomal protein MRPL12 interacts directly with mitochondrial RNA polymerase to modulate mitochondrial gene expression. J. Biol. Chem. 2007, 282, 12610–12618. [Google Scholar] [CrossRef]
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926. [Google Scholar] [CrossRef]
- Enriquez, J.A.; Fernandez-Silva, P.; Perez-Martos, A.; Lopez-Perez, M.J.; Montoya, J. The synthesis of mRNA in isolated mitochondria can be maintained for several hours and is inhibited by high levels of ATP. Eur. J. Biochem. 1996, 237, 601–610. [Google Scholar] [CrossRef]
- Ghosh, S.; Sengupta, S.; Scaria, V. Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 2014, 18, 58–62. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.; Rots, M.G. Mitochondrial epigenetics: An overlooked layer of regulation? Trends Genet. 2015, 31, 353–356. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Kim, S.; Farr, C.L.; Schaefer, K.T.; Randolph, K.M.; Tainer, J.A.; Kaguni, L.S. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J. Mol. Biol. 2006, 358, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Curth, U.; Urbanke, C.; Kang, C. Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 A resolution. Nat. Struct. Biol. 1997, 4, 153–157. [Google Scholar] [CrossRef]
- Wanrooij, S.; Fuste, J.M.; Farge, G.; Shi, Y.; Gustafsson, C.M.; Falkenberg, M. Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11122–11127. [Google Scholar] [CrossRef]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5′ -> 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed]
- Miralles Fuste, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, O.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Nadalutti, C.A.; Motori, E.; Sommerville, E.W.; Gorman, G.S.; Basu, S.; Hoberg, E.; Turnbull, D.M.; Chinnery, P.F.; Larsson, N.G.; et al. Topoisomerase 3alpha Is Required for Decatenation and Segregation of Human mtDNA. Mol. Cell 2018, 69, 9–23.e6. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Simsek, D.; Jasin, M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 2011, 10, 3636–3644. [Google Scholar] [CrossRef] [PubMed]
- Lakshmipathy, U.; Campbell, C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 2001, 29, 668–676. [Google Scholar] [CrossRef]
- Ruhanen, H.; Ushakov, K.; Yasukawa, T. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim. Biophys. Acta 2011, 1813, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol. Cell 2017, 65, 527–538.e526. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Cecconi, C.; Tkachuk, A.N.; Bustamante, C.; Clayton, D.A. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005, 19, 2466–2476. [Google Scholar] [CrossRef]
- Yasukawa, T.; Yang, M.Y.; Jacobs, H.T.; Holt, I.J. A bidirectional origin of replication maps to the major noncoding region of human mitochondrial DNA. Mol. Cell 2005, 18, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Bowmaker, M.; Reyes, A.; Vergani, L.; Angeli, P.; Gringeri, E.; Jacobs, H.T.; Holt, I.J. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell 2002, 111, 495–505. [Google Scholar] [CrossRef]
- Holt, I.J.; Lorimer, H.E.; Jacobs, H.T. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 2000, 100, 515–524. [Google Scholar] [CrossRef]
- Bowmaker, M.; Yang, M.Y.; Yasukawa, T.; Reyes, A.; Jacobs, H.T.; Huberman, J.A.; Holt, I.J. Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J. Biol. Chem. 2003, 278, 50961–50969. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects of mitochondrial DNA replication. J. Child Neurol. 2014, 29, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; McCullough, A.K.; Lloyd, R.S. Regulation of DNA glycosylases and their role in limiting disease. Free Radic. Res. 2012, 46, 460–478. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H. Oxidative DNA damage in disease–insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagen. 2014, 55, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Ozawa, T. Strand asymmetry in human mitochondrial DNA mutations. Genomics 1994, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Gissi, C.; Pesole, G.; Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998, 15, 957–966. [Google Scholar] [CrossRef]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. (Landmark Ed.) 2017, 22, 920–943. [Google Scholar]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Briede, J.J.; Jennen, D.G.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.; de Kok, T.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef]
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. (Clifton, NJ) 2009, 554, 165–181. [Google Scholar]
- Lambert, A.J.; Brand, M.D. Research on mitochondria and aging, 2006–2007. Aging Cell 2007, 6, 417–420. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.; Gonzalez-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.E.; Tran, K.; Smith, C.C.; McDonald, M.; Shejwalkar, P.; Hara, K. The Role of the Nrf2/ARE Antioxidant System in Preventing Cardiovascular Diseases. Diseases 2016, 4, 34. [Google Scholar] [CrossRef]
- Slocinska, M.; Barylski, J.; Jarmuszkiewicz, W. Uncoupling proteins of invertebrates: A review. IUBMB Life 2016, 68, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.J.; Van Houten, B. Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator of hydrogen peroxide in human fibroblasts. Mutat. Res. 1997, 385, 139–149. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Van Houten, B.; Jin, G.F.; Conklin, C.A.; Godley, B.F. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 1999, 68, 765–772. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Alexeyev, M.F.; Ledoux, S.P.; Wilson, G.L. Mitochondrial DNA and aging. Clin. Sci. (Lond. Engl.) 2004, 107, 355–364. [Google Scholar] [CrossRef]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. (Landmark Ed.) 2016, 21, 42–54. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Wilson, D.M.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair 2008, 7, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M., 3rd; Bohr, V.A. Repair of persistent strand breaks in the mitochondrial genome. Mech. Ageing Dev. 2012, 133, 169–175. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, 0181687. [Google Scholar] [CrossRef] [PubMed]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL-1 deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef]
- Thameem, F.; Puppala, S.; Lehman, D.M.; Stern, M.P.; Blangero, J.; Abboud, H.E.; Duggirala, R.; Habib, S.L. The Ser(326)Cys polymorphism of 8-oxoguanine glycosylase 1 (OGG1) is associated with Type 2 diabetes in Mexican Americans. Hum Hered 2010, 70, 97–101. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef]
- Parker, A.R.; Eshleman, J.R. Human MutY: Gene structure, protein functions and interactions, and role in carcinogenesis. Cell. Mol. Life Sci. 2003, 60, 2064–2083. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Polynucleotide kinase: A versatile molecule makes a clean break. Structure (Lond. Engl.) 2002, 10, 1151–1152. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Nakabeppu, Y.; Furuichi, M.; Sekiguchi, M. Regulation of expression of the human MTH1 gene encoding 8-oxo-dGTPase. Alternative splicing of transcription products. J. Biol. Chem. 1997, 272, 17843–17850. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926–4936. [Google Scholar] [CrossRef]
- Rolseth, V.; Runden-Pran, E.; Luna, L.; McMurray, C.; Bjoras, M.; Ottersen, O.P. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair 2008, 7, 1578–1588. [Google Scholar] [CrossRef]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef] [PubMed]
- Karahalil, B.; Hogue, B.A.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Sklar, J. Augmented expression of a human gene for 8-oxoguanine DNA glycosylase (MutM) in B lymphocytes of the dark zone in lymph node germinal centers. J. Exp. Med. 1997, 186, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar] [CrossRef]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Kajitani, K.; Yamaguchi, H.; Dan, Y.; Furuichi, M.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J. Neurosci. 2006, 26, 1688–1698. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Ohta, E.; Abolhassani, N. MTH1 as a nucleotide pool sanitizing enzyme: Friend or foe? Free Radic. Biol. Med. 2017, 107, 151–158. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001, 477, 59–70. [Google Scholar] [CrossRef]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Chakravarthy, S.; Longley, M.J.; Copeland, W.C.; Prakash, A. The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair 2018, 65, 11–19. [Google Scholar]
- Takao, M.; Kanno, S.; Shiromoto, T.; Hasegawa, R.; Ide, H.; Ikeda, S.; Sarker, A.H.; Seki, S.; Xing, J.Z.; Le, X.C.; et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002, 21, 3486–3493. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M.; Hegde, M.L.; Chatterjee, A.; Hegde, P.M.; Szczesny, B.; Banerjee, D.; Boldogh, I.; Gao, R.; Falkenberg, M.; Gustafsson, C.M.; et al. Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 2012, 287, 2819–2829. [Google Scholar] [CrossRef] [PubMed]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Thornley, N.P.; Grishko, V.I.; LeDoux, S.P.; Wilson, G.L. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 2006, 55, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; Solodushko, V.A.; Wilson, G.L.; Rachek, L.I. Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology 2012, 153, 92–100. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Schuler, A.M.; Chen, J.; Alvarez, D.F.; Eide, L.; Ledoux, S.P.; Wilson, G.L.; Rachek, L.I. Alteration of mitochondrial function and insulin sensitivity in primary mouse skeletal muscle cells isolated from transgenic and knockout mice: Role of ogg1. Endocrinology 2013, 154, 2640–2649. [Google Scholar] [CrossRef]
- Rachek, L.I.; Grishko, V.I.; Musiyenko, S.I.; Kelley, M.R.; LeDoux, S.P.; Wilson, G.L. Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. J. Biol. Chem. 2002, 277, 44932–44937. [Google Scholar] [CrossRef] [PubMed]
- Souza-Pinto, N.C.; Croteau, D.L.; Hudson, E.K.; Hansford, R.G.; Bohr, V.A. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999, 27, 1935–1942. [Google Scholar] [CrossRef]
- Lia, D.; Reyes, A.; de Melo Campos, J.T.A.; Piolot, T.; Baijer, J.; Radicella, J.P.; Campalans, A. Mitochondrial maintenance under oxidative stress depends on mitochondrially localised alpha-OGG1. J. Cell Sci. 2018, 131, jcs213538. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Hogue, B.A.; Bohr, V.A. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radic. Biol. Med. 2001, 30, 916–923. [Google Scholar] [CrossRef]
- Imai, K.; Sarker, A.H.; Akiyama, K.; Ikeda, S.; Yao, M.; Tsutsui, K.; Shohmori, T.; Seki, S. Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 1998, 222, 287–295. [Google Scholar] [CrossRef]
- Luna, L.; Bjoras, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000, 460, 95–104. [Google Scholar] [CrossRef]
- Ikeda, S.; Kohmoto, T.; Tabata, R.; Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair 2002, 1, 847–854. [Google Scholar] [CrossRef]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004, 32, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Wang, C.; Hu, Z.; Greeley, G.H.; Makalowski, W.; Hellmich, H.L.; Englander, E.W. Hypoxia induces mitochondrial DNA damage and stimulates expression of a DNA repair enzyme, the Escherichia coli MutY DNA glycosylase homolog (MYH), in vivo, in the rat brain. J. Neurochem. 2002, 80, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef]
- Yoshimura, D.; Sakumi, K.; Ohno, M.; Sakai, Y.; Furuichi, M.; Iwai, S.; Nakabeppu, Y. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. Biol. Chem. 2003, 278, 37965–37973. [Google Scholar] [CrossRef]
- Stojkovic, G.; Makarova, A.V.; Wanrooij, P.H.; Forslund, J.; Burgers, P.M.; Wanrooij, S. Oxidative DNA damage stalls the human mitochondrial replisome. Sci. Rep. 2016, 6, 28942. [Google Scholar] [CrossRef]
- Hanes, J.W.; Thal, D.M.; Johnson, K.A. Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J. Biol. Chem. 2006, 281, 36241–36248. [Google Scholar] [CrossRef]
- Garcia-Gomez, S.; Reyes, A.; Martinez-Jimenez, M.I.; Chocron, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Mendez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Posse, V.; Shahzad, S.; Falkenberg, M.; Hallberg, B.M.; Gustafsson, C.M. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015, 43, 2615–2624. [Google Scholar] [CrossRef]
- Nakanishi, N.; Fukuoh, A.; Kang, D.; Iwai, S.; Kuraoka, I. Effects of DNA lesions on the transcription reaction of mitochondrial RNA polymerase: Implications for bypass RNA synthesis on oxidative DNA lesions. Mutagenesis 2013, 28, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Maeda, L.S.; Kolodner, R.D.; Hanawalt, P.C. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair 2004, 3, 483–494. [Google Scholar] [CrossRef]
- Charlet-Berguerand, N.; Feuerhahn, S.; Kong, S.E.; Ziserman, H.; Conaway, J.W.; Conaway, R.; Egly, J.M. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006, 25, 5481–5491. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, I.; Suzuki, K.; Ito, S.; Hayashida, M.; Kwei, J.S.; Ikegami, T.; Handa, H.; Nakabeppu, Y.; Tanaka, K. RNA polymerase II bypasses 8-oxoguanine in the presence of transcription elongation factor TFIIS. DNA Repair 2007, 6, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Bautista-Nino, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef] [PubMed]
- Moehrle, B.M.; Geiger, H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol. 2016, 44, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 2005, 33, D611–D613. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Crippa, B.L.; Leon, E.; Calhoun, A.; Lowichik, A.; Pasquali, M.; Longo, N. Biochemical abnormalities in Pearson syndrome. Am. J. Med. Genet. Part A 2015, 3, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Saldana-Martinez, A.; Munoz, M.L.; Perez-Ramirez, G.; Montiel-Sosa, J.F.; Montoya, J.; Emperador, S.; Ruiz-Pesini, E.; Cuevas-Covarrubias, S.; Lopez-Valdez, J.; Ramirez, R.G. Whole sequence of the mitochondrial DNA genome of Kearns Sayre Syndrome patients: Identification of deletions and variants. Gene 2018, 688, 171–181. [Google Scholar] [CrossRef]
- Kim, U.S.; Jurkute, N.; Yu-Wai-Man, P. Leber Hereditary Optic Neuropathy-Light at the End of the Tunnel? Asia Pac. J. Ophthalmol. 2018, 7, 242–245. [Google Scholar] [CrossRef]
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondrial disorders. Epileptic Disord. 2016, 18, 94–102. [Google Scholar]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment strategies for inherited optic neuropathies: Past, present and future. Eye (Lond. Engl.) 2014, 28, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy—Therapeutic Challenges and Early Promise. Taiwan J. Ophthalmol. 2011, 1, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Szuhai, K.; Ouweland, J.; Dirks, R.; Lemaitre, M.; Truffert, J.; Janssen, G.; Tanke, H.; Holme, E.; Maassen, J.; Raap, A. Simultaneous A8344G heteroplasmy and mitochondrial DNA copy number quantification in myoclonus epilepsy and ragged-red fibers (MERRF) syndrome by a multiplex molecular beacon based real-time fluorescence PCR. Nucleic Acids Res. 2001, 29, E13. [Google Scholar] [CrossRef]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta 2015, 1847, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Salas, A.; Yao, Y.G.; Macaulay, V.; Vega, A.; Carracedo, A.; Bandelt, H.J. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med. 2005, 2, e296. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef]
- Horton, T.M.; Petros, J.A.; Heddi, A.; Shoffner, J.; Kaufman, A.E.; Graham, S.D., Jr.; Gramlich, T.; Wallace, D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosom. Cancer 1996, 15, 95–101. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Tse, K.H.; Herrup, K. Re-imagining Alzheimer’s disease—The diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 2017, 143, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.; Czarny, P.; Toma, M.; Jurkowska, N.; Sliwinska, A.; Drzewoski, J.; Bachurska, A.; Szemraj, J.; Maes, M.; Berk, M.; et al. Associations between DNA Damage, DNA Base Excision Repair Gene Variability and Alzheimer’s Disease Risk. Dement. Geriatr. Cogn. Disord. 2016, 41, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjoras, M.; Eide, L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci. 2011, 31, 9746–9751. [Google Scholar] [CrossRef]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Sakumi, K.; Castillo, E.; Sheng, Z.; Oka, S.; Nakabeppu, Y. 8-Oxoguanine accumulation in mitochondrial DNA causes mitochondrial dysfunction and impairs neuritogenesis in cultured adult mouse cortical neurons under oxidative conditions. Sci. Rep. 2016, 6, 22086. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5, 0012037. [Google Scholar] [CrossRef]
- Ikebe, S.; Tanaka, M.; Ohno, K.; Sato, W.; Hattori, K.; Kondo, T.; Mizuno, Y.; Ozawa, T. Increase of deleted mitochondrial DNA in the striatum in Parkinson’s disease and senescence. Biochem. Biophys. Res. Commun. 1990, 170, 1044–1048. [Google Scholar] [CrossRef]
- Simon, D.K.; Pulst, S.M.; Sutton, J.P.; Browne, S.E.; Beal, M.F.; Johns, D.R. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 1999, 53, 1787–1793. [Google Scholar] [CrossRef]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor’s Highlight: Base Excision Repair Variants and Pesticide Exposure Increase Parkinson’s Disease Risk. Toxicol. Sci. 2017, 158, 188–198. [Google Scholar] [CrossRef]
- Cornetta, T.; Patrono, C.; Terrenato, I.; De Nigris, F.; Bentivoglio, A.R.; Testa, A.; Palma, V.; Poggioli, T.; Padua, L.; Cozzi, R. Epidemiological, clinical, and molecular study of a cohort of Italian Parkinson disease patients: Association with glutathione-S-transferase and DNA repair gene polymorphisms. Cell. Mol. Neurobiol. 2013, 33, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Fukae, J.; Mizuno, Y.; Hattori, N. Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 2007, 7, 58–62. [Google Scholar] [CrossRef]
- Fukae, J.; Takanashi, M.; Kubo, S.; Nishioka, K.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Coppede, F.; Ceravolo, R.; Migheli, F.; Fanucchi, F.; Frosini, D.; Siciliano, G.; Bonuccelli, U.; Migliore, L. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson’s disease. Neurosci. Lett. 2010, 473, 248–251. [Google Scholar] [CrossRef]
- Skarpengland, T.; Holm, S.; Scheffler, K.; Gregersen, I.; Dahl, T.B.; Suganthan, R.; Segers, F.M.; Ostlie, I.; Otten, J.J.; Luna, L.; et al. Neil3-dependent base excision repair regulates lipid metabolism and prevents atheroslerosis in Apoe-deficient mice. Sci. Rep. 2016, 6, 28337. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, K.; Rachek, L.; You, P.; Rowe, A.D.; Wang, W.; Kusnierczyk, A.; Kittelsen, L.; Bjoras, M.; Eide, L. 8-oxoguanine DNA glycosylase (Ogg1) controls hepatic gluconeogenesis. DNA Repair 2018, 61, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; Musiyenko, S.I.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS ONE 2013, 8, e54059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, C.; Spencer, H.J.; Zuo, C.; Higuchi, M.; Ranganathan, G.; Kern, P.A.; Chou, M.W.; Huang, Q.; Szczesny, B.; et al. Obesity and hepatosteatosis in mice with enhanced oxidative DNA damage processing in mitochondria. Am. J. Pathol. 2011, 178, 1715–1727. [Google Scholar] [CrossRef]
- Hara, M.; Nakamura, K.; Nanri, H.; Nishida, Y.; Hishida, A.; Kawai, S.; Hamajima, N.; Kita, Y.; Suzuki, S.; Mantjoro, E.M.; et al. Associations between hOGG1 Ser326Cys polymorphism and increased body mass index and fasting glucose level in the Japanese general population. J. Epidemiol. 2014, 24, 379–384. [Google Scholar] [CrossRef]
- Corella, D.; Ramirez-Sabio, J.B.; Coltell, O.; Ortega-Azorin, C.; Estruch, R.; Martinez-Gonzalez, M.A.; Salas-Salvado, J.; Sorli, J.V.; Castaner, O.; Aros, F.; et al. Effects of the Ser326Cys Polymorphism in the DNA Repair OGG1 Gene on Cancer. J. Acad. Nutr. Diet. 2018, 118, 589–605. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Enzyme | Glycosylase Family | Tissue Specificity | Inferred or Confirmed Function(s) of Mitochondrial Isoform | Mitochondrial Localization Described |
|---|---|---|---|---|
| NEIL1 | Fpg/Nei Helix-two turns-helix | Liver, thymus, pancreas, brain [128,129,130] | Potential role in mediating metabolic syndrome in Neil1−/− mice [141]; Binding partner for mtSSB [142] | [142,143] |
| NEIL2 | Fpg/Nei Helix-two turns-helix | Testes and skeletal muscle [131] | Removal of oxidized bases from mitochondrial genome [144] | [144] |
| OGG1 (1a) | Helix-hairpin- helix | Thymus, testis, intestine, brain, and germinal center of B cells [130,133,135,145] | Role in protecting against oxidative stress [146,147,148,149]; Prevention of metabolic syndrome [116] | [150,151,152] |
| NTH1 | Helix-hairpin- helix | Heart, brain [130,132,153] | Unknown; potentially compensated for by NEIL1 activity | Mouse isoform is exclusively mitochondrial; human isoform is thought to be exclusively nuclear [154,155] |
| MUTY | Helix-hairpin- helix | Thymus, intestine, heart, lung [156] | Potentially involved in repair of hypoxia induced damage in brain [157] | [156,158] |
| MTH1 | Oxidized purine nucleoside triphosphatase | Thymus, testis, embryonic tissues [127] | Protection from oxidative damage in models of Parkinson’s disease [137] | [136,138,159] |
| Disease | Clinical Manifestations | Location |
|---|---|---|
| Leber Hereditary Optic Neuropathy (LHON) | Bilateral, painless subacute visual failure | Mutations in MT-ND1, MT-ND4 or MT-ND6 gene [179,180] |
| Myoclonic Epilepsy with Ragged Red Fibers (MERRF) | Myoclonus epilepsy, ataxia, muscle weakness and dementia | Mutations in MT-TK encoding tRNA lysine (tRNALys). A-to-G transition at nucleotide 8344 [178,181] |
| Pearson’s syndrome | Sideroblastic anemia and exocrine pancreas dysfunction | Mitochondrial DNA deletion [174,182] |
| Kearns-Sayre Syndrome (KSS) | Pigmentary retinopathy | Mitochondrial DNA deletion [176] |
| Mitochondrial encephalopathy, lactic acidosis and strokelike episodes (MELAS) | Mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes. Other features include headache, seizures, muscle weakness | Point mutation in the tRNA leucineUUR gene of mitochondrial DNA. A to G transition at nucleotide 3243 [173] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. https://doi.org/10.3390/cells8020100
Sharma P, Sampath H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells. 2019; 8(2):100. https://doi.org/10.3390/cells8020100
Chicago/Turabian StyleSharma, Priyanka, and Harini Sampath. 2019. "Mitochondrial DNA Integrity: Role in Health and Disease" Cells 8, no. 2: 100. https://doi.org/10.3390/cells8020100
APA StyleSharma, P., & Sampath, H. (2019). Mitochondrial DNA Integrity: Role in Health and Disease. Cells, 8(2), 100. https://doi.org/10.3390/cells8020100