Yeast to Study Human Purine Metabolism Diseases



Abstract
1. Introduction
2. Purine Metabolism in Human and Yeast: Similarities and Differences
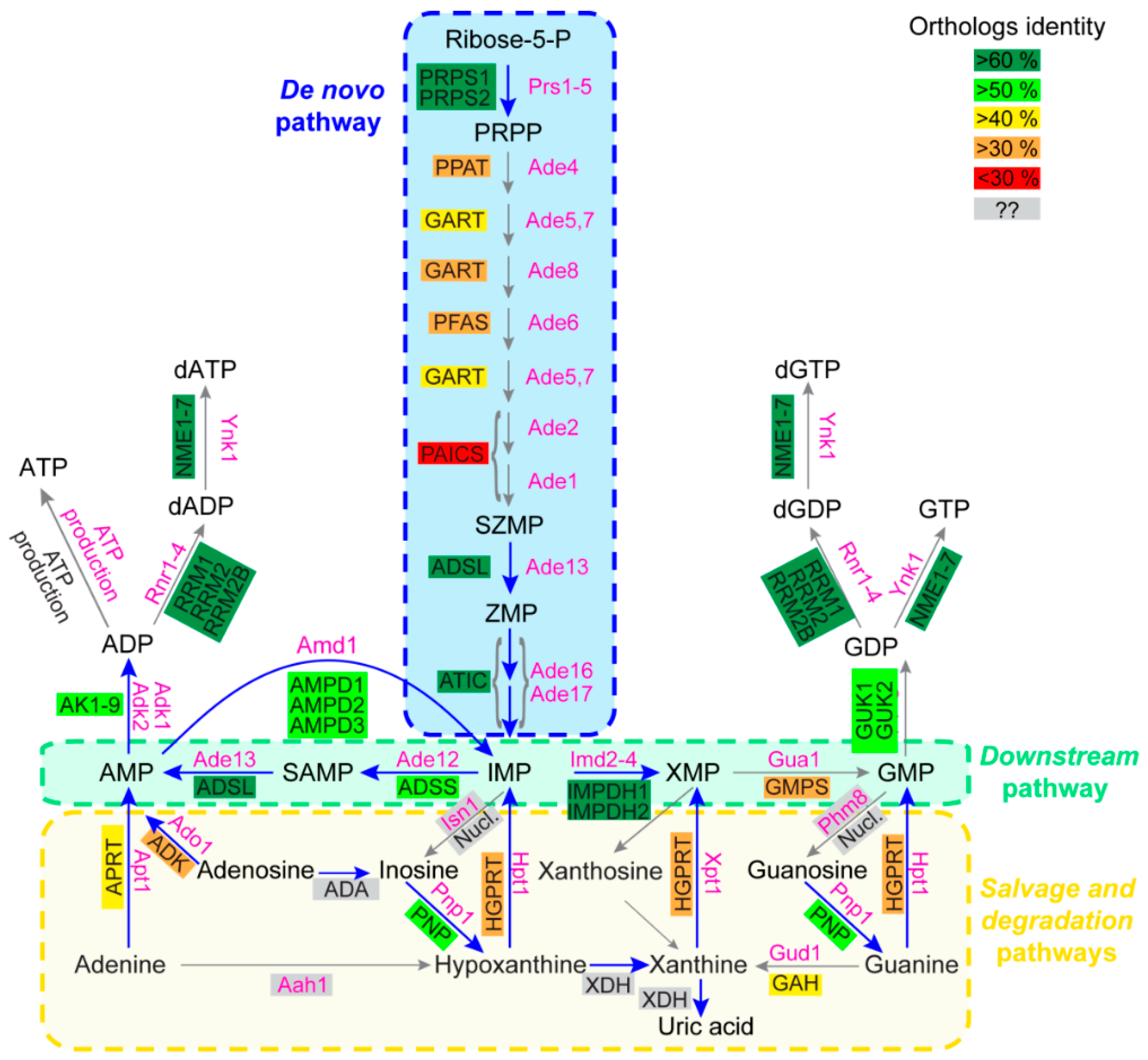
Purine Metabolic Pathways: Functions Are Generally Conserved but Protein Sequences Can Diverge
3. Yeast: A Model to Tackle Complex Purine-Associated Diseases
- Important metabolites may not be synthesized, or synthesized insufficiently
- Metabolites, which are accumulated due to an increased synthesis or the lack of metabolizing enzymes, can be toxic because they interfere with other processes (this could be particularly true for metabolites which have physiological regulatory properties)
- A metabolic unbalance can occur, which is often due to a mix of the first two hypotheses
3.1. AMP-Deaminase (AMPD2) Deficiency Associated with Pontocerebellar Hypoplasia Results in Defective ATP/GTP Balance
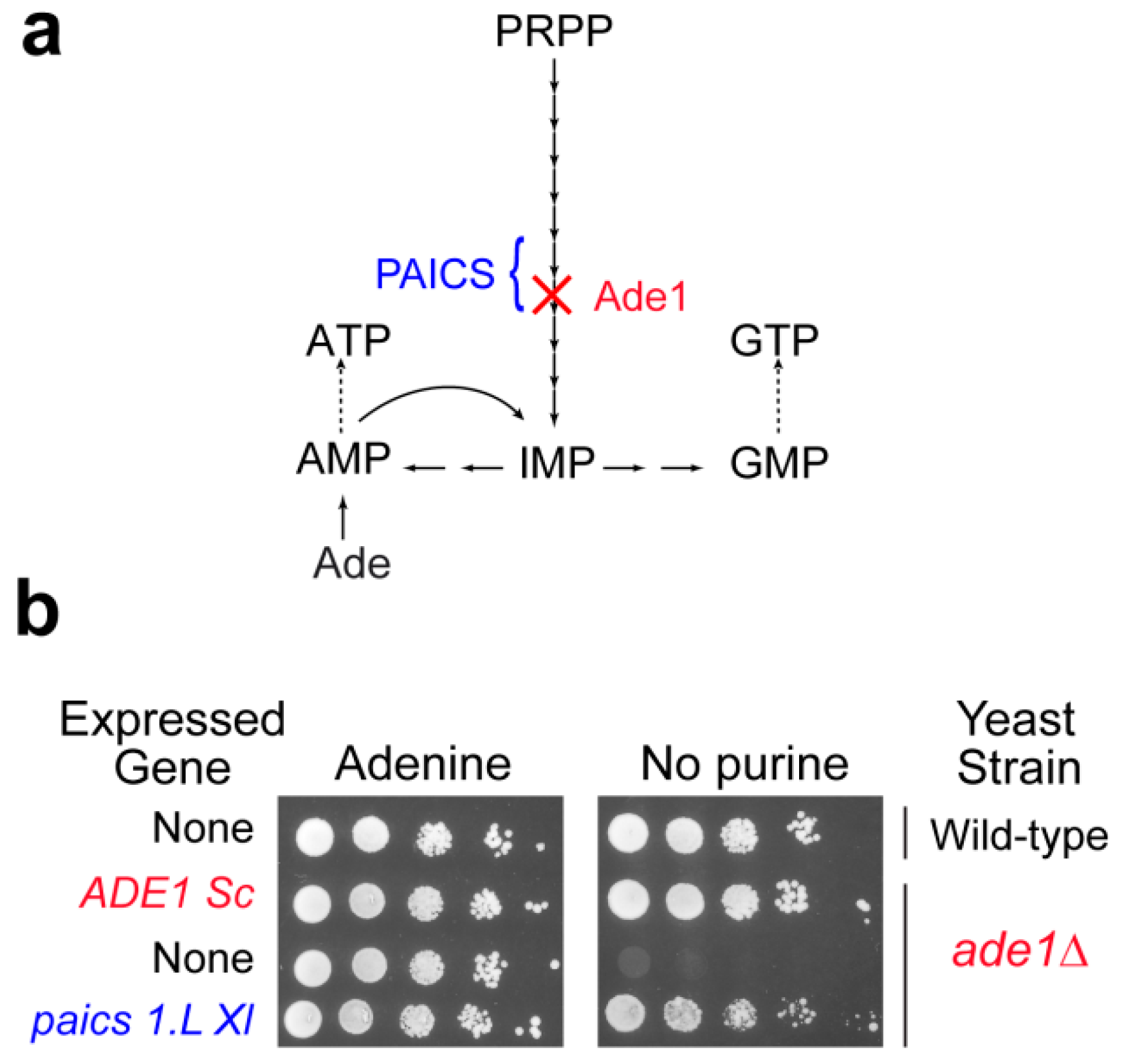
3.2. Deficiencies in the Purine De Novo Pathway: Toxic Accumulation of Metabolic Intermediates?
4. Yeast as a Tool to Functionally Validate Orthologous Genes from Other Model Organisms
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Protein Name | ORF Name | Enzymatic Activities |
|---|---|---|
| Aah1 | YNL141W | Adenine deaminase |
| Ade1 | YAR015W | Phosphoribosyl aminoimidazole succinocarboxamide synthetase |
| Ade12 | YNL220W | Adenylosuccinate synthase |
| Ade13 | YLR359W | Adenylosuccinate lyase |
| Ade16 | YLR028C | AICAR transformylase and IMP cyclohydrolase |
| Ade17 | YMR120C | AICAR transformylase and IMP cyclohydrolase |
| Ade2 | YOR128C | Phosphoribosylaminoimidazole carboxylase |
| Ade4 | YMR300C | Phosphoribosylpyrophosphate amidotransferase |
| Ade5,7 | YGL234W | Aminoimidazole ribonucleotide synthetase and glycinamide ribotide synthetase |
| Ade6 | YGR061C | Formylglycinamidine-ribonucleotide synthetase |
| Ade8 | YDR408C | Phosphoribosyl-glycinamide transformylase |
| Adk1 | YDR226W | Adenylate kinase |
| Adk2 | YER170W | Adenylate kinase |
| Ado1 | YJR105W | Adenosine kinase |
| Amd1 | YML035C | AMP deaminase |
| Apt1 | YML022W | Adenine phosphoribosyltransferase |
| Gua1 | YMR217W | GMP synthase |
| Gud1 | YDL238C | Guanine deaminase |
| Guk1 | YDR454C | Guanylate kinase |
| Hpt1 | YDR399W | Hypoxanthine-guanine phosphoribosyltransferase |
| Imd2 | YHR216W | Inosine monophosphate dehydrogenase |
| Imd3 | YLR432W | Inosine monophosphate dehydrogenase |
| Imd4 | YML056C | Inosine monophosphate dehydrogenase |
| Isn1 | YOR155C | Inosine monophosphate specific nucleotidase |
| Phm8 | YER037W | GMP, UMP and CMP nucleotidase |
| Pnp1 | YLR209C | Purine nucleoside phosphorylase |
| Prs1 | YKL181W | PRPP synthetase subunit |
| Prs2 | YER099C | PRPP synthetase subunit |
| Prs3 | YHL011C | PRPP synthetase subunit |
| Prs4 | YBL068W | PRPP synthetase subunit |
| Prs5 | YOL061W | PRPP synthetase subunit |
| Rnr1 | YER070W | Large subunit of ribonucleotide-diphosphate reductase |
| Rnr2 | YJL026W | Small subunit of ribonucleotide-diphosphate reductase |
| Rnr3 | YIL066C | Large subunit of ribonucleotide-diphosphate reductase |
| Rnr4 | YGR180C | Small subunit of ribonucleotide-diphosphate reductase |
| Ynk1 | YKL067W | Nucleoside diphosphate kinase |
| Protein Name | Gene Location | Enzymatic Activity |
|---|---|---|
| ADA | 20q13.12 | Adenosine deaminase |
| ADK | 10q22 | Adenosine kinase |
| ADSL | 22q13.1 | Adenylosuccinate lyase |
| ADSS | 1q44 | Adenylosuccinate synthase |
| AK1 | 9q34.11 | Adenylate kinase |
| AK2 | 1p35.1 | Adenylate kinase |
| AK3 | 9p24.1 | Adenylate kinase |
| AK4 | 1p31.3 | Adenylate kinase |
| AK5 | 1p31.1 | Adenylate kinase |
| AK6 | 5q13.2 | Adenylate kinase |
| AK7 | 14q32.2 | Adenylate kinase |
| AK8 | 9q34.13 | Adenylate kinase |
| AK9 | 6q21 | Adenylate kinase |
| AMPD1 | 1p13.2 | Adenosine monophosphate deaminase |
| AMPD2 | 1p13.3 | Adenosine monophosphate deaminase |
| AMPD3 | 11p15.4 | Adenosine monophosphate deaminase |
| APRT | 16q24.3 | Adenine phosphoribosyltransferase |
| ATIC | 2q35 | Amino-Imidazole CarboxAmide Ribonucleotide transformylase and IMP cyclohydrolase |
| GAH | 9q21.13 | Guanine deaminase |
| GART | 21q22.11 | Phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase and phosphoribosylaminoimidazole synthetase |
| GMPS | 3q25.31 | Guanine monophosphate synthase |
| GUK1 | 1q42.13 | Guanylate kinase |
| GUK2 | 1q32.1-q42 | Guanylate kinase |
| HPRT1 | Xq26.1 | Hypoxanthine phosphoribosyltransferase |
| IMPDH1 | 7q32.1 | Inosine monophosphate dehydrogenase |
| IMPDH2 | 3p21.31 | Inosine monophosphate dehydrogenase |
| NME1 | 17q21.33 | Nucleoside diphosphate kinase |
| NME2 | 17q21.33 | Nucleoside diphosphate kinase |
| NME3 | 16p13.3 | Nucleoside diphosphate kinase |
| NME4 | 16p13.3 | Nucleoside diphosphate kinase |
| NME5 | 5q31.2 | Nucleoside diphosphate kinase |
| NME6 | 3p21.31 | Nucleoside diphosphate kinase |
| NME7 | 1q24.2 | Nucleoside diphosphate kinase |
| PAICS | 4q12 | Phosphoribosylaminoimidazole carboxylase and phosphoribosylaminoimidazolesuccinocarboxamide synthetase |
| PFAS | 17p13.1 | Phosphoribosylformylglycinamidine synthase |
| PNP | 14q11.2 | Purine nucleoside phosphorylase |
| PPAT | 4q12 | Phosphoribosyl pyrophosphate amidotransferase |
| PRPS1 | Xq22.3 | Phosphoribosyl pyrophosphate synthetase 1 |
| PRPS2 | Xp22.2 | Phosphoribosyl pyrophosphate synthetase 2 |
| RRM1 | 11p15.4 | Ribonucleotide reductase catalytic subunit |
| RRM2 | 2p25.1 | Ribonucleotide reductase regulatory subunit |
| RRM2B | 8q22.3 | Ribonucleotide reductase regulatory subunit |
| XDH | 2p23.1 | Xanthine dehydrogenase/oxidase |
| Yeast Protein | Human Protein | Identity (%) | Similarity (%) | Protein Coverage (%) |
|---|---|---|---|---|
| Rnr2 | RMM2 | 69 | 81 | 90 |
| Rnr3 | RMM1 | 66 | 84 | 87 |
| Ade13 | ADSL | 64 | 79 | 97 |
| Prs3 | PRPS2 | 61 | 78 | 79 |
| Ynk1 | NME2 | 61 | 80 | 97 |
| Imd2 | IMPD2 | 61 | 78 | 98 |
| Prs2 | PRPS1 | 60 | 75 | 78 |
| Ade16 | ATIC | 60 | 75 | 99 |
| Ade17 | ATIC | 60 | 75 | 99 |
| Adk1 | AK2 | 58 | 73 | 98 |
| Ade12 | ADSS | 52 | 68 | 99 |
| Pnp1 | PNP | 51 | 68 | 85 |
| Guk1 | GUK1 | 51 | 70 | 99 |
| Amd1 | AMPD2 | 50 | 66 | 74 |
| Ade5,7 | GART | 45 | 64 | 99 |
| Apt1 | APRT | 44 | 64 | 96 |
| Gud1 | GAH | 42 | 49 | 99 |
| Ado1 | ADK1 | 36 | 57 | 99 |
| Gua1 | GMPS | 36 | 54 | 100 |
| Ade4 | ATASE/PPAT | 34 | 51 | 92 |
| Ade6 | PFAS | 33 | 49 | 99 |
| Hpt1 | HGPRT | 33 | 46 | 33 |
| Ade8 | GART | 32 | 52 | 92 |
| Ade2 | PAICS | 27 | 45 | 99 |
| Ade1 | PAICS | 22 | 41 | 100 |
References
- Ljungdahl, P.O.; Daignan-Fornier, B. Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 2012, 190, 885–929. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Sabina, R.L.; van den Berghe, G. Metabolic disorders of purine metabolism affecting the nervous system. Handb. Clin. Neurol. 2013, 113, 1827–1836. [Google Scholar]
- Jurecka, A. Inborn errors of purine and pyrimidine metabolism. J. Inherit. Metab. Dis. 2009, 32, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Nyhan, W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005, 86, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Seegmiller, J.E.; Rosenbloom, F.M.; Kelley, W.N. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science 1967, 155, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, H.A.; Duley, J.A.; Fairbanks, L.D.; McBride, M.B. When to investigate for purine and pyrimidine disorders. Introduction and review of clinical and laboratory indications. J. Inherit. Metab. Dis. 1997, 20, 214–226. [Google Scholar] [CrossRef]
- Van den Berghe, G.; Vincent, F.; Marie, S. Disorders of purine and pyrimidine metabolism. In Inborn Metabolic Diseases, 4th ed.; Springer: Berlin, Germany, 2006; pp. 433–449. [Google Scholar]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef]
- Keller, K.E.; Doctor, Z.M.; Dwyer, Z.W.; Lee, Y.S. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol. Cell 2014, 53, 700–709. [Google Scholar] [CrossRef]
- Keller, K.E.; Tan, I.S.; Lee, Y.S. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 2012, 338, 1069–1072. [Google Scholar] [CrossRef]
- Pinson, B.; Vaur, S.; Sagot, I.; Coulpier, F.; Lemoine, S.; Daignan-Fornier, B. Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 2009, 23, 1399–1407. [Google Scholar] [CrossRef]
- Rebora, K.; Desmoucelles, C.; Borne, F.; Pinson, B.; Daignan-Fornier, B. Yeast AMP pathway genes respond to adenine through regulated synthesis of a metabolic intermediate. Mol. Cell Biol. 2001, 21, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Rebora, K.; Laloo, B.; Daignan-Fornier, B. Revisiting purine-histidine cross-pathway regulation in Saccharomyces cerevisiae: A central role for a small molecule. Genetics 2005, 170, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Letisse, F.; Absalan, F.; Lu, W.; Kuznetsova, E.; Brown, G. Nucleotide degradation and ribose salvage in yeast. Mol. Syst. Biol. 2013, 9, 665. [Google Scholar] [CrossRef] [PubMed]
- Itoh, R.; Saint-Marc, C.; Chaignepain, S.; Katahira, R.; Schmitter, J.M.; Daignan-Fornier, B. The yeast ISN1 (YOR155c) gene encodes a new type of IMP-specific 5’-nucleotidase. BMC Biochem. 2003, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.; Spychala, J. Mammalian 5’-nucleotidases. J. Biol. Chem. 2003, 278, 46195–46198. [Google Scholar] [CrossRef] [PubMed]
- Akeson, A.L.; Wiginton, D.A.; Hutton, J.J. Normal and mutant human adenosine deaminase genes. J. Cell Biochem. 1989, 39, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ribard, C.; Rochet, M.; Labedan, B.; Daignan-Fornier, B.; Alzari, P.; Scazzocchio, C. Sub-families of alpha/beta barrel enzymes: A new adenine deaminase family. J. Mol. Biol. 2003, 334, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- Pinson, B.; Napias, C.; Chevallier, J.; Van den Broek, P.J.; Brethes, D. Characterization of the Saccharomyces cerevisiae cytosine transporter using energizable plasma membrane vesicles. J. Biol. Chem. 1997, 272, 28918–28924. [Google Scholar] [CrossRef]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Asp. Med. 2013, 34, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Armitt, S.; Woods, R.A. Purine-excreting mutants of Saccharomyces cerevisiae. I. Isolation and genetic analysis. Genet. Res. 1970, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, K.; Konrad, M.; Daignan-Fornier, B. Yeast GMP kinase mutants constitutively express AMP biosynthesis genes by phenocopying a hypoxanthine-guanine phosphoribosyltransferase defect. Genetics 2000, 156, 953–961. [Google Scholar] [PubMed]
- Jaeken, J.; van den Berghe, G. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 1984, 2, 1058–1061. [Google Scholar] [PubMed]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.A.; Farrar, G.J.; Kenna, P.; Humphries, M.M.; Sheils, D.M.; Kumar-Singh, R. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat. Genet. 1993, 4, 54–58. [Google Scholar] [CrossRef]
- Bowne, S.J.; Sullivan, L.S.; Mortimer, S.E.; Hedstrom, L.; Zhu, J.; Spellicy, C.J. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Hansson, O.; Liden, S.; Nilsson, K. Familial ataxic diplegia with deficient cellular immunity. A new clinical entity. Acta Paediatr. Scand. 1970, 59, 545–550. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518. [Google Scholar] [CrossRef]
- Park, J.; Hyun, Y.S.; Kim, Y.J.; Nam, S.H.; Kim, S.H.; Hong, Y.B. Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy. J. Clin. Neurol. 2013, 9, 283–288. [Google Scholar] [CrossRef]
- Synofzik, M.; Muller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wodl, S. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: Evidence from a family with a novel PRPS1 mutation. Orphanet J. Rare Dis. 2014, 9, 24. [Google Scholar] [CrossRef]
- Becker, M.A.; Puig, J.G.; Mateos, F.A.; Jimenez, M.L.; Kim, M.; Simmonds, H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988, 85, 383–390. [Google Scholar] [CrossRef]
- Saint-Marc, C.; Pinson, B.; Coulpier, F.; Jourdren, L.; Lisova, O.; Daignan-Fornier, B. Phenotypic consequences of purine nucleotide imbalance in Saccharomyces cerevisiae. Genetics 2009, 183, 529–538. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Rathbone, M.P.; Middlemiss, P.J.; Gysbers, J.W.; Andrew, C.; Herman, M.A.; Reed, J.K. Trophic effects of purines in neurons and glial cells. Prog. Neurobiol. 1999, 59, 663–690. [Google Scholar] [CrossRef]
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymanska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Wadman, S.K.; Duran, M.; van Sprang, F.J.; Beemer, F.A.; Holl, R.A. Adenylosuccinase deficiency: An inborn error of purine nucleotide synthesis. Eur. J. Pediatr. 1988, 148, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Race, V.; Marie, S.; Vincent, M.F.; Van den Berghe, G. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 2000, 9, 2159–2165. [Google Scholar] [CrossRef]
- Jurecka, A.; Zikanova, M.; Tylki-Szymanska, A.; Krijt, J.; Bogdanska, A.; Gradowska, W. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2008, 94, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant adsl enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Douillet, D.C.; Pinson, B.; Ceschin, J.; Hurlimann, H.C.; Saint-Marc, C.; Laporte, D. Metabolomics and proteomics identify the toxic form and the associated cellular binding targets of the anti-proliferative drug AICAR. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Le Dantec, A.; Brassier, A.; Jais, J.P.; Ledroit, M.; Cahu, J. New biomarkers for early diagnosis of Lesch-Nyhan disease revealed by metabolic analysis on a large cohort of patients. Orphanet J. Rare Dis. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Sidi, Y.; Mitchell, B.S. Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J. Clin. Investig. 1985, 76, 2416–2419. [Google Scholar] [CrossRef] [PubMed]
- Hurlimann, H.C.; Laloo, B.; Simon-Kayser, B.; Saint-Marc, C.; Coulpier, F.; Lemoine, S. Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J. Biol. Chem. 2011, 286, 30994–31002. [Google Scholar] [CrossRef] [PubMed]
- Ceschin, J.; Hurlimann, H.C.; Saint-Marc, C.; Albrecht, D.; Violo, T.; Moenner, M. Disruption of Nucleotide Homeostasis by the Antiproliferative Drug 5-Aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside Monophosphate (AICAR). J. Biol. Chem. 2015, 290, 23947–23959. [Google Scholar] [CrossRef]
- Lopez, J.M. Is ZMP the toxic metabolite in Lesch-Nyhan disease? Med. Hypotheses 2008, 71, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 39582–39593. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Williams, B.R.; Siegel, J.J.; Amon, A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011, 144, 499–512. [Google Scholar] [CrossRef]
- Ly, P.; Kim, S.B.; Kaisani, A.A.; Marian, G.; Wright, W.E.; Shay, J.W. Aneuploid human colonic epithelial cells are sensitive to AICAR-induced growth inhibition through EGFR degradation. Oncogene 2013, 32, 3139–3146. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; Gonzalez-Barca, E.; Terol, M.J.; Levy, V. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (CLL): A multicenter phase I/II study. Cancer Chemother. Pharmacol. 2013, 71, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef]
- Theodoropoulou, S.; Brodowska, K.; Kayama, M.; Morizane, Y.; Miller, J.W.; Gragoudas, E.S. Aminoimidazole carboxamide ribonucleotide (AICAR) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis. PLoS ONE 2013, 8, e52852. [Google Scholar] [CrossRef] [PubMed]
- Ceschin, J.; Saint-Marc, C.; Laporte, J.; Labriet, A.; Philippe, C.; Moenner, M. Identification of yeast and human 5-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside (AICAr) transporters. J. Biol. Chem. 2014, 289, 16844–16854. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.; Ceschin, J.; Dompierre, J.; Gueniot, F.; Pinson, B.; Daignan-Fornier, B. Chemo-Genetic Interactions Between Histone Modification and the Antiproliferation Drug AICAR Are Conserved in Yeast and Humans. Genetics 2016, 204, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.; Hurlimann, H.C.; Ceschin, J.; Saint-Marc, C.; Pinson, B.; Daignan-Fornier, B. Multiple chemo-genetic interactions between a toxic metabolite and the ubiquitin pathway in yeast. Curr. Genet. 2018, 64, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Daignan-Fornier, B.; Pinson, B. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5’-Monophosphate (AICAR), a Highly Conserved Purine Intermediate with Multiple Effects. Metabolites 2012, 2, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Desmoucelles, C.; Pinson, B.; Saint-Marc, C.; Daignan-Fornier, B. Screening the yeast “disruptome” for mutants affecting resistance to the immunosuppressive drug, mycophenolic acid. J. Biol. Chem. 2002, 277, 27036–27044. [Google Scholar] [CrossRef]
- Kuehn, M.R.; et al. A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 1987, 326, 295–298. [Google Scholar] [CrossRef]
- Ng, A.; Uribe, R.A.; Yieh, L.; Nuckels, R.; Gross, J.M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 2009, 136, 2601–2611. [Google Scholar] [CrossRef]
- Marsac, R.; Pinson, B.; Saint-Marc, C.; Olmedo, M.; Artal-Sanz, M.; Daignan-Fornier, B.; Gomes, J.-E. Adenylosuccinate Lyase activity in the Purine recycling pathway is essential for developmental timing, germline maintenance and muscle integrity in C. elegans. bioRxiv 2018. [Google Scholar] [CrossRef]
- Huyet, J.; Ozeir, M.; Burgevin, M.C.; Pinson, B.; Chesney, F.; Remy, J.M. Structural Insights into the Forward and Reverse Enzymatic Reactions in Human Adenine Phosphoribosyltransferase. Cell Chem. Biol. 2018, 25, 666–676. [Google Scholar] [CrossRef]
- The Saccharomyces Genome Database. Available online: https://www.yeastgenome.org (accessed on 16 January 2019).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 16 January 2019).
- ProteomeTM Platform. Available online: https://portal.genexplain.com (accessed on 16 January 2019).


| Pathology | Enzymatic Defect | Gene Name (Location) | Phenotype MIM Number | Inheritance | Locus MIM Number | Reference |
|---|---|---|---|---|---|---|
| ADSL deficiency | Loss of function | ADSL (22q13.1) | 103050 | Autosomal recessive | 608222 | [24] |
| AICA-Ribosiduria | Loss of function | ATIC (2q35) | 608688 | Autosomal recessive | 601731 | [25] |
| Retinitis pigmentosa 10 | Loss of function | IMPDH1 (7q32.1) | 180105 | Autosomal dominant | [26] | |
| Leber Congenital Amaurosis 11 | Loss of function | IMPDH1 (7q32.1) | 613837 | ? | 146690 | [27] |
| PNP deficiency | PNP (14q11.2) | 613179 | Autosomal recessive | 164050 | [28] | |
| Arts syndrome | Loss of function | PRPS1 (Xq22.3) | 311835 | X-linked recessive | 311850 | [29] |
| Charcot-Marie-Tooth disease, X-linked recessive, 5 | Loss of function | PRPS1 (Xq22.3) | 311070 | X-linked recessive | 311850 | [30] |
| Deafness, X-linked 1 | Loss of function | PRPS1 (Xq22.3) | 304500 | X-linked | 311850 | [31] |
| Hyperuricemia, PRPS-related | Gain of function | PRPS1 (Xq22.3) | 300661 | X-linked recessive | 311850 | [32] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daignan-Fornier, B.; Pinson, B. Yeast to Study Human Purine Metabolism Diseases. Cells 2019, 8, 67. https://doi.org/10.3390/cells8010067
Daignan-Fornier B, Pinson B. Yeast to Study Human Purine Metabolism Diseases. Cells. 2019; 8(1):67. https://doi.org/10.3390/cells8010067
Chicago/Turabian StyleDaignan-Fornier, Bertrand, and Benoît Pinson. 2019. "Yeast to Study Human Purine Metabolism Diseases" Cells 8, no. 1: 67. https://doi.org/10.3390/cells8010067
APA StyleDaignan-Fornier, B., & Pinson, B. (2019). Yeast to Study Human Purine Metabolism Diseases. Cells, 8(1), 67. https://doi.org/10.3390/cells8010067