Recent Insights into NCL Protein Function Using the Model Organism Dictyostelium discoideum

Abstract
1. Neuronal Ceroid Lipofuscinosis
2. Studying the Functions of NCL Proteins Using the Model Organism Dictyostelium Discoideum
3. Using Dictyostelium to Study CLN2 Disease
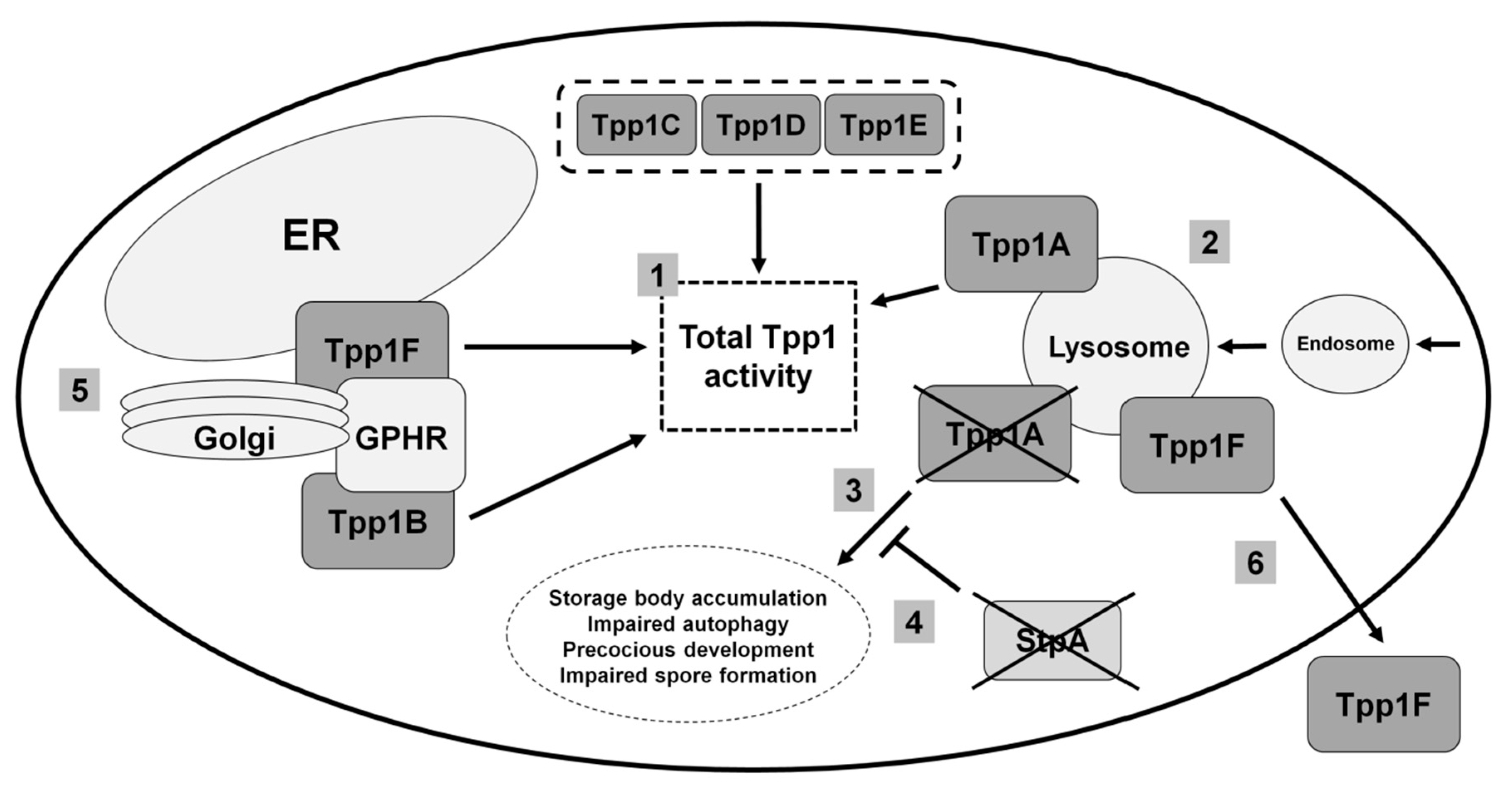
3.1. Human TPP1
3.2. Loss of the Lysosomal Enzyme Tpp1A Impairs Autophagy and Multicellular Development in Dictyostelium
3.3. Tpp1B and Tpp1F Interact with the Golgi pH Regulator in Dictyostelium
4. Using Dictyostelium to Study CLN3 Disease
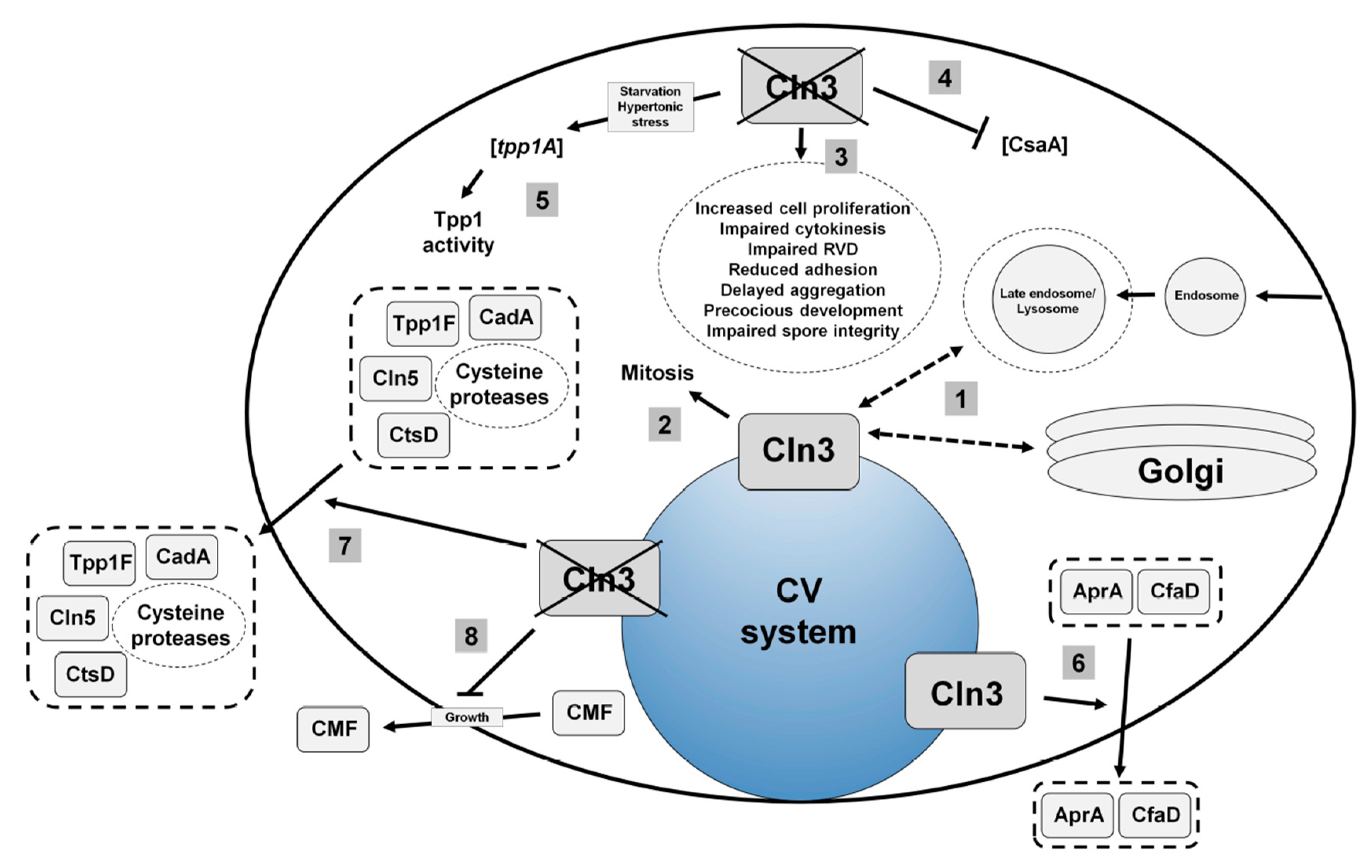
4.1. Human CLN3
4.2. Loss of Cln3 Causes Pleiotropic Effects in Dictyostelium that are Consistent with its Localization to the Contractile Vacuole System
4.3. Cln3 Regulates Osmoregulation in Dictyostelium
4.4. Cln3 Regulates Protein Secretion in Dictyostelium
5. Using Dictyostelium to Study CLN5 Disease
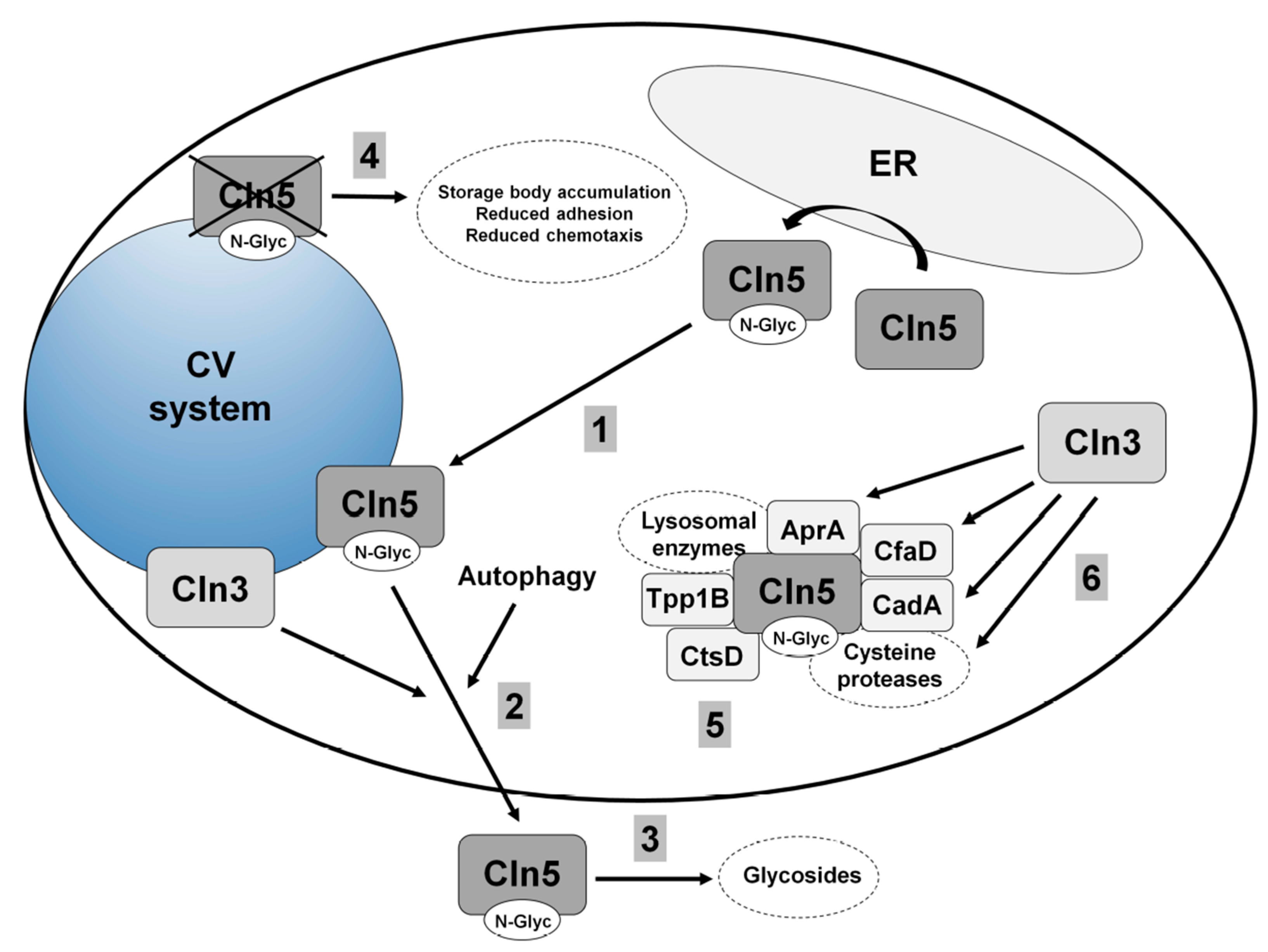
5.1. Human CLN5
5.2. Cln5 is Secreted and Functions as a Glycoside Hydrolase in Dictyostelium
5.3. Loss of Cln5 Impairs Adhesion and Chemotaxis during the Early Stages of Dictyostelium Development
6. Using Dictyostelium to Study the Molecular Networking of NCL Proteins
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AprA | autocrine proliferation repressor A |
| CadA | cell adhesion molecule A |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| CfaD | counting factor-associated protein D |
| CLN | ceroid lipofuscinosis neuronal |
| CMF | conditioned media factor |
| CsaA | contact site A |
| CtsD | cathepsin D |
| CV | contractile vacuole |
| ER | endoplasmic reticulum |
| GPHR | Golgi pH regulator |
| NCL | neuronal ceroid lipofuscinosis |
| REMI | restriction enzyme-mediated integration |
| RVD | regulatory volume decrease |
| SCAR7 | spinocerebellar ataxia 7 |
| SPPL | signal peptide peptidase-like |
| StpA | suppressor of Tpp1 A |
| TPP1 | tripeptidyl peptidase 1 |
References
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL neuropathology. Biochim. Biophys. Acta 2015, 1852, 2262–2266. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Kohlschütter, A.; Mink, J.; Simonati, A.; Williams, R. NCL diseases—Clinical perspectives. Biochim. Biophys. Acta 2013, 1832, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Holthaus, S.; Tammen, I.; Tear, G.; Russell, C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta 2013, 1832, 1842–1865. [Google Scholar] [CrossRef] [PubMed]
- Müller-Taubenberger, A.; Kortholt, A.; Eichinger, L. Simple system—Substantial share: The use of Dictyostelium in cell biology and molecular medicine. Eur. J. Cell Biol. 2013, 92, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Using the social amoeba Dictyostelium to study the functions of proteins linked to neuronal ceroid lipofuscinosis. J. Biomed. Sci. 2016, 23, 83. [Google Scholar] [CrossRef]
- Eichinger, L.; Pachebat, J.A.; Glöckner, G.; Rajandream, M.-A.; Sucgang, R.; Berriman, M.; Song, J.; Olsen, R.; Szafranski, K.; Xu, Q.; et al. The genome of social amoeba Dictyostelium discoideum. Nature 2005, 435, 43–57. [Google Scholar] [CrossRef]
- Mathavarajah, S.; Flores, A.; Huber, R.J. Dictyostelium discoideum: A model system for cell and developmental biology. Curr. Protoc. Essent. Lab. Tech. 2017, 15, 14.1.1–14.1.19. [Google Scholar]
- Faix, J.; Linkner, J.; Nordholz, B.; Platt, J.L.; Liao, X.H.; Kimmel, A.R. The application of the Cre-loxP system for generating multiple knock-out and knock-in targeted loci. Methods Mol. Biol. 2013, 983, 249–267. [Google Scholar]
- Sekine, R.; Kawata, T.; Muramoto, T. CRISPR/Cas9 mediated targeting of multiple genes in Dictyostelium. Sci. Rep. 2018, 8, 8471. [Google Scholar] [CrossRef] [PubMed]
- Terbach, N.; Shah, R.; Kelemen, R.; Klein, P.S.; Gordienko, D.; Brown, N.A.; Wilkinson, C.J.; Williams, R.S. Identifying an uptake mechanism for the antiepileptic and bipolar disorder treatment valproic acid using the simple biomedical model Dictyostelium. J. Cell Sci. 2011, 124, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Walker, M.C.; Williams, R.S. Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid. Neurobiol. Dis. 2014, 62, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Alexander, H. Lead genetic studies in Dictyostelium discoideum and translational studies in human cells demonstrate that sphingolipids are key regulators of sensitivity to cisplatin and other anticancer drugs. Semin. Cell Dev. Biol. 2011, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Meyer, I.; Kuhnert, O.; Gräf, R. Functional analyses of lissencephaly-related proteins in Dictyostelium. Semin. Cell Dev. Biol. 2011, 22, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Maniak, M. Dictyostelium as a model for human lysosomal and trafficking diseases. Semin. Cell Dev. Biol. 2011, 22, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Myre, M.A. Clues to γ-secretase, huntingtin and Hirano body normal function using the model organism Dictyostelium discoideum. J. Biomed. Sci. 2012, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C.; Williams, R.S. The search for better epilepsy treatments: From slime mould to coconuts. Biochem. Soc. Trans. 2013, 41, 1625–1628. [Google Scholar] [CrossRef]
- Annesley, S.J.; Chen, S.; Francione, L.M.; Sanislav, O.; Chavan, A.J.; Farah, C.; De Piazza, S.W.; Storey, C.L.; Ilievska, J.; Fernando, S.G.; et al. Dictyostelium, a microbial model for brain disease. Biochim. Biophys. Acta 2014, 1840, 1413–1432. [Google Scholar] [CrossRef]
- Malinovska, L.; Alberti, S. Protein misfolding in Dictyostelium: Using a freak of nature to gain insight into a universal problem. Prion 2015, 9, 339–346. [Google Scholar] [CrossRef]
- Sun, Y.; Almomani, R.; Breedveld, G.J.; Santen, G.W.; Aten, E.; Lefeber, D.J.; Hoff, J.I.; Brusse, E.; Verheijen, F.W.; Verdijk, R.M.; et al. Autosomal recessive spinocerebellar ataxia 7 (SCAR7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN2 disease). Hum. Mutat. 2013, 34, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Donnelly, R.J.; Lackland, H.; Liu, C.G.; Sohar, I.; Pullarkat, R.K.; Lobel, P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 1997, 277, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Donet, J.M.; Cárcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS ONE 2013, 8, e55526. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Gomer, R.H. Partial genetic suppression of a loss-of-function mutant of the neuronal ceroid lipofuscinosis-associated protease TPP1 in Dictyostelium discoideum. Dis. Models Mech. 2015, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.; Müller, R.; Gaßen, B.; Wehrstedt, R.; Fey, P.; Karow, M.A.; Eichinger, L.; Glöckner, G.; Noegel, A.A. A tripeptidyl peptidase 1 is a binding partner of the Golgi pH regulator (GPHR) in Dictyostelium. Dis. Models Mech. 2017, 10, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Li, S. Oxysterol-binding proteins: Sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog. Lipid Res. 2013, 52, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Sima, N.; Li, R.; Huang, W.; Xu, M.; Beers, J.; Zou, J.; Titus, S.; Ottinger, E.A.; Marugan, J.J.; Xie, X.; et al. Neural stem cells for disease modeling and evaluation of therapeutics for infantile (CLN1/PPT1) and late infantile (CLN2/TPP1) neuronal ceroid lipofuscinoses. Orphanet J. Rare Dis. 2018, 13, 54. [Google Scholar] [CrossRef]
- Schultz, M.L.; Tecedor, L.; Lysenko, E.; Ramachandran, S.; Stein, C.S.; Davidson, B.L. Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo. Neurobiol. Dis. 2018, 115, 182–193. [Google Scholar] [CrossRef]
- Maeda, Y.; Ide, T.; Koike, M.; Uchiyama, Y.; Kinoshita, T. GPHR is a novel anion channel critical for acidification and functions of the Golgi apparatus. Nat. Cell Biol. 2008, 10, 1135–1145. [Google Scholar] [CrossRef]
- Charroux, B.; Royet, J. Mutations in the Drosophila ortholog of the vertebrate Golgi pH regulator (GPHR) protein disturb endoplasmic reticulum and Golgi organization and affect systemic growth. Biol. Open 2014, 3, 72–80. [Google Scholar] [CrossRef]
- Deckstein, J.; van Appeldorn, J.; Tsangarides, M.; Yiannakou, K.; Müller, R.; Stumpf, M.; Sukumaran, S.K.; Eichinger, L.; Noegel, A.A.; Riyahi, T.Y. The Dictyostelium discoideum GPHR ortholog is an endoplasmic reticulum and Golgi protein with roles during development. Eukaryot. Cell 2015, 14, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Loss of Cln3 impacts protein secretion in the social amoeba Dictyostelium. Cell. Signal. 2017, 35, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Rot, G.; Parikh, A.; Curk, T.; Kuspa, A.; Shaulsky, G.; Zupan, B. dictyExpress: A Dictyostelium discoideum gene expression database with an explorative data analysis web-based interface. BMC Bioinform. 2009, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Staropoli, J.F. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin. Lipidol. 2012, 7, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, E.; Petcherski, A.; Ramos-Moreno, J.; Ruonala, M.O. FRET-assisted determination of CLN3 membrane topology. PLoS ONE 2014, 9, e102593. [Google Scholar] [CrossRef] [PubMed]
- Codlin, S.; Haines, R.L.; Burden, J.J.; Mole, S.E. Btn1 affects cytokinesis and cell-wall deposition by independent mechanisms, one of which is linked to dysregulation of vacuole pH. J. Cell Sci. 2008, 121, 2860–2870. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.S.; Yancey, P.H.; Martins, I.; Sigmund, R.D.; Stokes, J.B.; Davidson, B.L. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am. J. Physiol. Cell Physiol. 2010, 298, C1388–C1400. [Google Scholar] [CrossRef]
- Getty, A.; Kovács, A.D.; Lengyel-Nelson, T.; Cardillo, A.; Hof, C.; Chan, C.H.; Pearce, D.A. Osmotic stress changes the expression and subcellular localization of the Batten disease protein CLN3. PLoS ONE 2013, 8, e66203. [Google Scholar] [CrossRef]
- Tecedor, L.; Stein, C.S.; Schultz, M.L.; Farwanah, H.; Sandhoff, K.; Davidson, B.L. CLN3 loss disturbs membrane microdomain properties and protein transport in brain endothelial cells. J. Neurosci. 2013, 33, 18065–18079. [Google Scholar] [CrossRef]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS ONE 2014, 9, e110544. [Google Scholar] [CrossRef]
- Fabritius, A.; Vesa, J.; Minye, H.M.; Nakano, I.; Kornblum, H.; Peltonen, L. Neuronal ceroid lipofuscinosis genes, CLN2, CLN3 and CLN5 are spatially and temporally co-expressed in a developing mouse brain. Exp. Mol. Pathol. 2014, 97, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Mao, D.; Che, J.; Han, S.; Zhao, H.; Zhu, Y.; Zhu, H. RNAi-mediated knockdown of the CLN3 gene inhibits proliferation and promotes apoptosis in drug-resistant ovarian cancer cells. Mol. Med. Rep. 2015, 12, 6635–6641. [Google Scholar] [CrossRef] [PubMed]
- Chandrachud, U.; Walker, M.W.; Simas, A.M.; Heetveld, S.; Petcherski, A.; Klein, M.; Oh, H.; Wolf, P.; Zhao, W.N.; Norton, S.; et al. Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J. Biol. Chem. 2015, 290, 14361–14380. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Song, K.D.; Lee, H.K.; Yi, S.; Lee, Y.S.; Heo, T.H.; Jun, H.S.; Kim, S.J. Fibrates inhibit the apoptosis of Batten disease lymphoblast cells via autophagy recovery and regulation of mitochondrial membrane potential. In Vitro Cell. Dev. Biol. Anim. 2016, 52, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Aberrant adhesion impacts early development in a Dictyostelium model for juvenile neuronal ceroid lipofuscinosis. Cell Adhes. Migr. 2017, 11, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Parviainen, L.; Dihanich, S.; Anderson, G.W.; Wong, A.M.; Brooks, H.R.; Abeti, R.; Rezaie, P.; Lalli, G.; Pope, S.; Heales, S.J.; et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 2017, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Tannous, A.; Sohar, I.; Wiseman, J.A.; Zheng, H.; Qian, M.; Zhao, C.; Xin, W.; Barone, R.; Sims, K.B.; et al. Proteomic analysis of brain and cerebrospinal fluid from the three major forms of neuronal ceroid lipofuscinosis reveals potential biomarkers. J. Proteome Res. 2017, 16, 3787–3804. [Google Scholar] [CrossRef]
- Mathavarajah, S.; McLaren, M.D.; Huber, R.J. Cln3 function is linked to osmoregulation in a Dictyostelium model of Batten disease. Biochim. Biophys. Acta 2018, 1864, 3559–3573. [Google Scholar] [CrossRef]
- Du, F.; Edwards, K.; Shen, Z.; Sun, B.; De Lozanne, A.; Briggs, S.; Firtel, R.A. Regulation of contractile vacuole formation and activity in Dictyostelium. EMBO J. 2008, 27, 2064–2076. [Google Scholar] [CrossRef]
- Sriskanthadevan, S.; Brar, S.K.; Manoharan, K.; Siu, C.H. Ca2+-calmodulin interacts with DdCAD-1 and promotes DdCAD-1 transport by contractile vacuoles in Dictyostelium cells. FEBS J. 2013, 280, 1795–1806. [Google Scholar] [CrossRef]
- Plattner, H. Contractile vacuole complex—Its expanding protein inventory. Int. Rev. Cell Mol. Biol. 2013, 306, 371–416. [Google Scholar]
- Bosch, M.E.; Kielian, T. Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities. J. Neurochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Maeno, E.; Shimizu, T.; Dezaki, K.; Wang, J.; Morishima, S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol. 2001, 532, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, M.; Puett, D. Inhibition of the degradation of receptor-bound human choriogonadotropin by lysosomotropic agents, protease inhibitors, and metabolic inhibitors. J. Biol. Chem. 1978, 253, 7832–7838. [Google Scholar] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO: TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Fukui, Y.; Inoué, S. Cell division in Dictyostelium with special emphasis on actomyosin organization in cytokinesis. Cell Motil. Cytoskeleton 1991, 18, 41–54. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, T.; Clarke, M. Calmodulin and the contractile vacuole complex in mitotic cells of Dictyostelium discoideum. J. Cell Sci. 1993, 104, 1119–1127. [Google Scholar]
- Neujahr, R.; Heizer, C.; Gerisch, G. Myosin II-independent processes in mitotic cells of Dictyostelium discoideum: Redistribution of the nuclei, re-arrangement of the actin system and formation of the cleavage furrow. J. Cell Sci. 1997, 110, 123–137. [Google Scholar]
- Wienke, D.C.; Knetsch, M.L.; Neuhaus, E.M.; Reedy, M.C.; Manstein, D.J. Disruption of a dynamin homologue affects endocytosis, organelle morphology, and cytokinesis in Dictyostelium discoideum. Mol. Biol. Cell 1999, 10, 225–243. [Google Scholar] [CrossRef]
- Rivero, F.; Illenberger, D.; Somesh, B.P.; Dislich, H.; Adam, N.; Meyer, A.K. Defects in cytokinesis, actin reorganization and the contractile vacuole in cells deficient in RhoGDI. EMBO J. 2002, 21, 4539–4549. [Google Scholar] [CrossRef]
- Gerald, N.J.; Siano, M.; De Lozanne, A. The Dictyostelium LvsA protein is localized on the contractile vacuole and is required for osmoregulation. Traffic 2002, 3, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Brock, D.A.; Gomer, R.H. A secreted factor represses cell proliferation in Dictyostelium. Development 2005, 132, 4553–4562. [Google Scholar] [CrossRef] [PubMed]
- Bakthavatsalam, D.; Brock, D.A.; Nikravan, N.N.; Houston, K.D.; Hatton, R.D.; Gomer, R.H. The secreted Dictyostelium protein CfaD is a chalone. J. Cell Sci. 2008, 121, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Gomer, R.H. A secreted protein is an endogenous chemorepellant in Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA 2012, 109, 10990–10995. [Google Scholar] [CrossRef] [PubMed]
- Gomer, R.H.; Yuen, I.S.; Firtel, R.A. A secreted 80 × 103 Mr protein mediates sensing of cell density and the onset of development in Dictyostelium. Development 1991, 112, 269–278. [Google Scholar] [PubMed]
- Carcel-Trullols, J.; Kovacs, A.D.; Pearce, D.A. Role of the lysosomal membrane protein, CLN3, in the regulation of cathepsin D activity. J. Cell. Biochem. 2017, 118, 3883–3890. [Google Scholar] [CrossRef]
- Cannelli, S.; Nardocci, N.; Cassandrini, D.; Morbin, M.; Aiello, C.; Bugiani, M.; Criscuolo, L.; Zara, F.; Striano, P.; Granata, T.; et al. Revelation of a novel CLN5 mutation in early juvenile neuronal ceroid lipofuscinosis. Neuropediatrics 2007, 38, 46–49. [Google Scholar] [CrossRef]
- Mancini, C.; Nassani, S.; Guo, Y.; Chen, Y.; Giorgio, E.; Brussino, A.; Di Gregorio, E.; Cavalieri, S.; Lo Buono, N.; Funaro, A.; et al. Adult-onset autosomal recessive ataxia associated with neuronal ceroid lipofuscinosis type 5 gene (CLN5) mutations. J. Neurol. 2015, 262, 173–178. [Google Scholar] [CrossRef]
- Savukoski, M.; Klockars, T.; Holmberg, V.; Santavuori, P.; Lander, E.S.; Peltonen, L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat. Genet. 1998, 19, 286–288. [Google Scholar] [CrossRef]
- Xin, W.; Mullen, T.E.; Kiely, R.; Min, J.; Feng, X.; Cao, Y.; O’Malley, L.; Shen, Y.; Chu-Shore, C.; Mole, S.E.; et al. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 2010, 74, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Santorelli, F.M.; Garavaglia, B.; Cardona, F.; Nardocci, N.; Bernardina, B.D.; Sartori, S.; Suppiej, A.; Bertini, E.; Claps, D.; Battini, R.; et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J. Rare Dis. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Williams, R.E.; Nardocci, N.; Laine, M.; Battini, R.; Schulz, A.; Garavaglia, B.; Moro, F.; Pezzini, F.; Santorelli, F.M. Phenotype and natural history of variant late infantile ceroid-lipofuscinosis 5. Dev. Med. Child Neurol. 2017, 59, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Isosomppi, J.; Vesa, J.; Jalanko, A.; Peltonen, L. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum. Mol. Genet. 2002, 11, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Moharir, A.; Peck, S.H.; Budden, T.; Lee, S.Y. The role of N-glycosylation in folding, trafficking, and functionality of lysosomal protein CLN5. PLoS ONE 2013, 8, e74299. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.M.; Hope, K.M.; Xu, J.B.; Mitchell, N.L.; Palmer, D.N. Inhibition of storage pathology in prenatal CLN5-deficient sheep neural cultures by lentiviral gene therapy. Neurobiol. Dis. 2014, 62, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Mathavarajah, S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell. Signal. 2018, 42, 236–248. [Google Scholar] [CrossRef]
- Larkin, H.; Ribeiro, M.G.; Lavoie, C. Topology and membrane anchoring of the lysosomal storage disease-related protein CLN5. Hum. Mutat. 2013, 34, 1688–1697. [Google Scholar] [CrossRef]
- De Silva, B.; Adams, J.; Lee, S.Y. Proteolytic processing of the neuronal ceroid lipofuscinosis related lysosomal protein CLN5. Exp. Cell Res. 2015, 338, 45–53. [Google Scholar] [CrossRef]
- Jules, F.; Sauvageau, E.; Dumaresq-doiron, K.; Mazzaferri, J.; Haug-Kröper, M.; Fluhrer, R.; Costantino, S.; Lefrancois, S. CLN5 is cleaved by members of the SPP/SPPL family to produce a mature soluble protein. Exp. Cell Res. 2017, 357, 40–50. [Google Scholar] [CrossRef]
- Mamo, A.; Jules, F.; Dumaresq-Doiron, K.; Costantino, S.; Lefrancois, S. The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Mol. Cell. Biol. 2012, 32, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Schmiedt, M.L.; Blom, T.; Blom, T.; Kopra, O.; Wong, A.; von Schantz-Fant, C.; Ikonen, E.; Kuronen, M.; Jauhiainen, M.; Cooper, J.D.; et al. Cln5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol. Dis. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Haddad, S.E.; Khoury, M.; Daoud, M.; Kantar, R.; Harati, H.; Mousallem, T.; Alzate, O.; Meyer, B.; Boustany, R. CLN5 and CLN8 protein association with ceramide synthase: Biochemical and proteomic approaches. Electrophoresis 2012, 33, 3798–3809. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.; Keksa-Goldsteine, V.; Ragauskas, S.; Kohlmann, P.; Singh, Y.; Savchenko, E.; Puranen, J.; Malm, T.; Kalesnykas, G.; Koistinaho, J.; et al. Retinal degeneration in a mouse model of CLN5 disease is associated with compromised autophagy. Sci. Rep. 2017, 7, 1597. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Feuerborn, M.; Molina, J.A.; Wilden, A.R.; Adhikari, B.; Budden, T.; Lee, S.Y. Autophagy-lysosome pathway alterations and alpha-synuclein up-regulation in the subtype of neuronal ceroid lipofuscinosis, CLN5 disease. Sci. Rep. 2019, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Mathavarajah, S. Secretion and function of Cln5 during the early stages of Dictyostelium development. Biochim. Biophys. Acta 2018, 1865, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, A.; Cardenal-Muñoz, E.; Dominguez, E.; Muñoz-Braceras, S.; Nuñez-Corcuera, B.; Phillips, B.A.; Tábara, L.C.; Xiong, Q.; Coria, R.; Eichinger, L.; et al. Autophagy in Dictyostelium: Mechanisms, regulation and disease in a simple biomedical model. Autophagy 2017, 13, 24–40. [Google Scholar] [CrossRef]
- Rote, K.V.; Rechsteiner, M. Degradation of microinjected proteins: Effects of lysosomotropic agents and inhibitors of autophagy. J. Cell. Physiol. 1983, 116, 103–110. [Google Scholar] [CrossRef]
- O’Day, D.H. Aggregation during sexual development in Dictyostelium discoideum. Can. J. Microbiol. 1979, 25, 1416–1426. [Google Scholar] [CrossRef]
- Schulz, A.; Dhar, S.; Rylova, S.; Dbaibo, G.; Alroy, J.; Hagel, C.; Artacho, I.; Kohlschütter, A.; Lin, S.; Boustany, R.M. Impaired cell adhesion and apoptosis in a novel CLN9 Batten disease variant. Ann. Neurol. 2004, 56, 342–350. [Google Scholar] [CrossRef]
- von Schantz, C.; Saharinen, J.; Kopra, O.; Cooper, J.D.; Gentile, M.; Hovatta, I.; Peltonen, L.; Jalanko, A. Brain gene expression profiles of Cln1 and Cln5 deficient mice unravels common molecular pathways underlying neuronal degeneration in NCL diseases. BMC Genom. 2008, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Persaud-Sawin, D.A.; Mousallem, T.; Wang, C.; Zucker, A.; Kominami, E.; Boustany, R.M. Neuronal ceroid lipofuscinosis: A common pathway? Pediatr. Res. 2007, 61, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Vesa, J.; Chin, M.H.; Oelgeschläger, K.; Isosomppi, J.; DellAngelica, E.C.; Jalanko, A.; Peltonen, L. Neuronal ceroid lipofuscinoses are connected at molecular level: Interaction of CLN5 protein with CLN2 and CLN3. Mol. Biol. Cell 2002, 13, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Lyly, A.; von Schantz, C.; Heine, C.; Schmiedt, M.L.; Sipilä, T.; Jalanko, A.; Kyttälä, A. Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biol. 2009, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Scifo, E.; Szwajda, A.; Dębski, J.; Uusi-Rauva, K.; Kesti, T.; Dadlez, M.; Gingras, A.C.; Tyynelä, J.; Baumann, M.H.; Jalanko, A.; et al. Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: A label-free quantitative proteomics approach. J. Proteome Res. 2013, 12, 2101–2115. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Schmiedt, M.L.; Wong, A.M.; Kyttälä, A.; Soronen, J.; Jauhiainen, M.; Tyynelä, J.; Cooper, J.D.; Jalanko, A. Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Dis. Models Mech. 2013, 6, 342–357. [Google Scholar] [CrossRef]
- Danyukova, T.; Ariunbat, K.; Thelen, M.; Brocke-Ahmadinejad, N.; Mole, S.E.; Storch, S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum. Mol. Genet. 2018, 27, 1711–1722. [Google Scholar] [PubMed]
- Journet, A.; Klein, G.; Brugière, S.; Vandenbrouck, Y.; Chapel, A.; Kieffer, S.; Bruley, C.; Masselon, C.; Aubry, L. Investigating the macropinocytic proteome of Dictyostelium amoebae by high-resolution mass spectrometry. Proteomics 2012, 12, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Xiu, X.; Jankovic, J. Genetic convergence of Parkinson’s disease and lysosomal storage disorders. Mol. Neurobiol. 2015, 51, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Dearborn, J.T.; Harmon, S.K.; Fowler, S.C.; O’Malley, K.L.; Taylor, G.T.; Sands, M.S.; Wozniak, D.F. Comprehensive functional characterization of murine infantile Batten disease including Parkinson-like behavior and dopaminergic markers. Sci. Rep. 2015, 5, 12752. [Google Scholar] [CrossRef]
- Qureshi, Y.H.; Patel, V.M.; Berman, D.E.; Kothiya, M.J.; Neufeld, J.L.; Vardarajan, B.; Tang, M.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; et al. An Alzheimer’s disease-linked loss-of-function CLN5 variant impairs cathepsin D maturation, consistent with a retromer trafficking defect. Mol. Cell. Biol. 2018, 38, e00011-18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dictyBase ID | Protein Names | Gene Names |
|---|---|---|
| DDB0231036 | Autocrine proliferation repressor protein A (PhoPQ-activated pathogenicity-related protein) | aprA DDB_G0281663 |
| DDB0215012 | Cathepsin D (Ddp44) | ctsD, catD DDB_G0279411 |
| DDB0214999 | Cysteine proteinase 4 | cprD, CP4 DDB_G0278721 |
| DDB0185092 | Cysteine proteinase 5 | cprE, CP5 DDB_G0272815 |
| DDB0215005 | Cysteine proteinase 7 | cprG, CP7 DDB_G0279187 |
| DDB0191134 | Elongation factor 1-alpha (EF-1-alpha) (50 kDa actin-binding protein) (ABP-50) | eef1a2, efaa2, efaAII DDB_G0269136 |
| DDB0233663 | Luminal-binding protein (BiP 2) | bip2 DDB_G0276445 |
| DDB0349243 | Uncharacterized protein | DDB_G0288563 |
| DDB0233868 | Uncharacterized protein, member of the peptidase S28 family of serine proteases, a group containing lysosomal Pro-X carboxypeptidase, dipeptidyl-peptidase II, and thymus-specific serine peptidase | DDB_G0289749 |
| DDB0238155 | Induced after Legionella infection Contains a putative N-terminal signal sequence; regulated by gskA and zakA; induced by Legionella pneumophila infection | iliA DDB_G0285615 |
| Phenotype | Tpp1a− | Cln3− | Cln5− |
|---|---|---|---|
| Increased cell proliferation | No | Yes | Not known |
| Impaired cytokinesis | Not known | Yes | Not known |
| Autofluorescent inclusions | Yes | Not known | Yes |
| Defects in osmoregulation | Not known | Yes | Not known |
| Aberrant protein secretion | Not known | Yes | Not known |
| Reduced adhesion | Not known | Yes | Yes |
| Function linked to autophagy | Yes | Not known | Yes |
| Precocious development | Yes | Yes | Not known |
| Impaired spore formation | Yes | Not known | Not known |
| Reduced spore viability/integrity | No | Yes | Not known |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLaren, M.D.; Mathavarajah, S.; Huber, R.J. Recent Insights into NCL Protein Function Using the Model Organism Dictyostelium discoideum. Cells 2019, 8, 115. https://doi.org/10.3390/cells8020115
McLaren MD, Mathavarajah S, Huber RJ. Recent Insights into NCL Protein Function Using the Model Organism Dictyostelium discoideum. Cells. 2019; 8(2):115. https://doi.org/10.3390/cells8020115
Chicago/Turabian StyleMcLaren, Meagan D., Sabateeshan Mathavarajah, and Robert J. Huber. 2019. "Recent Insights into NCL Protein Function Using the Model Organism Dictyostelium discoideum" Cells 8, no. 2: 115. https://doi.org/10.3390/cells8020115
APA StyleMcLaren, M. D., Mathavarajah, S., & Huber, R. J. (2019). Recent Insights into NCL Protein Function Using the Model Organism Dictyostelium discoideum. Cells, 8(2), 115. https://doi.org/10.3390/cells8020115