The Precision Control of Autophagic Flux and Vesicle Dynamics—A Micropattern Approach

 , and
, and 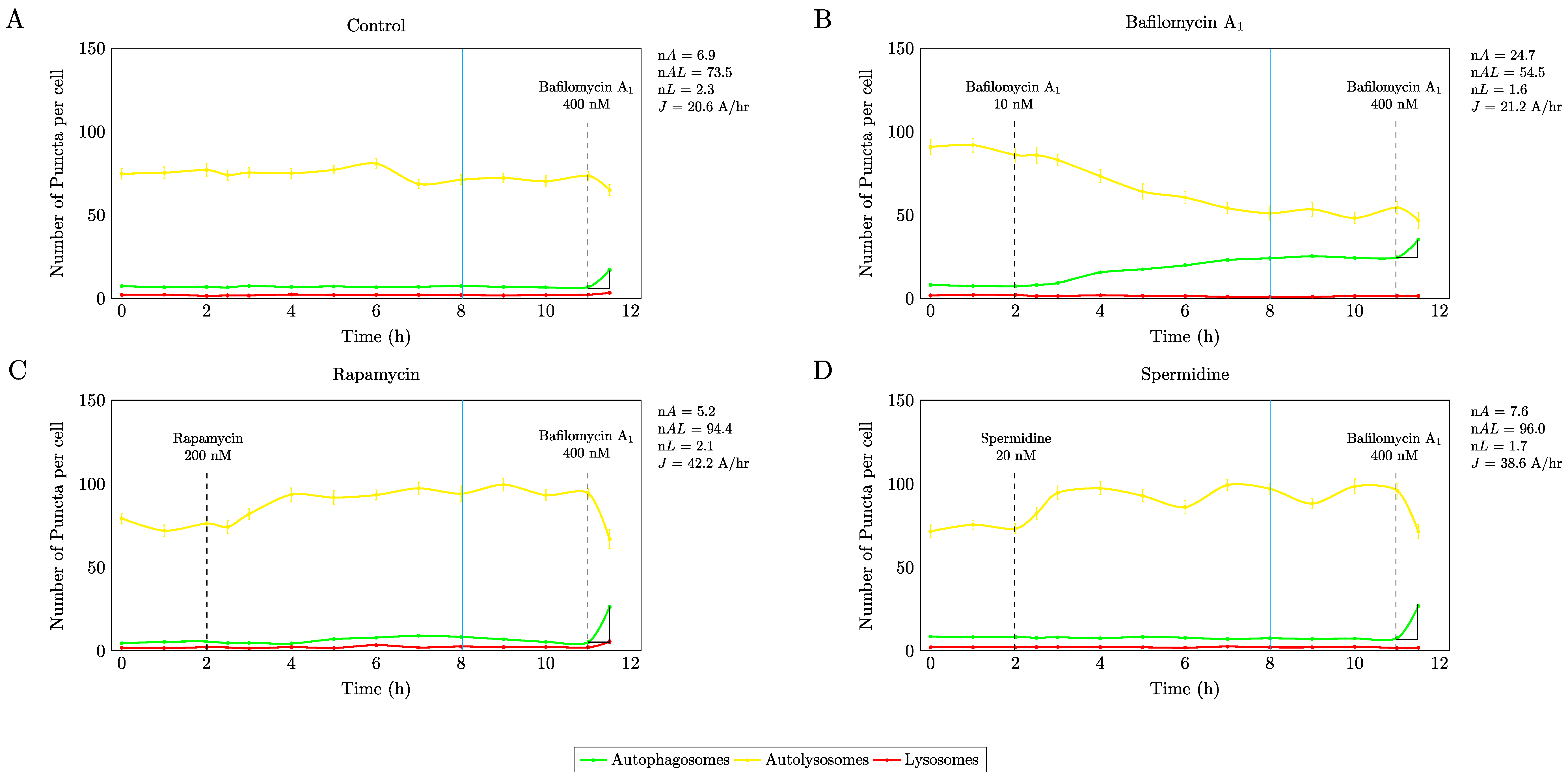{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
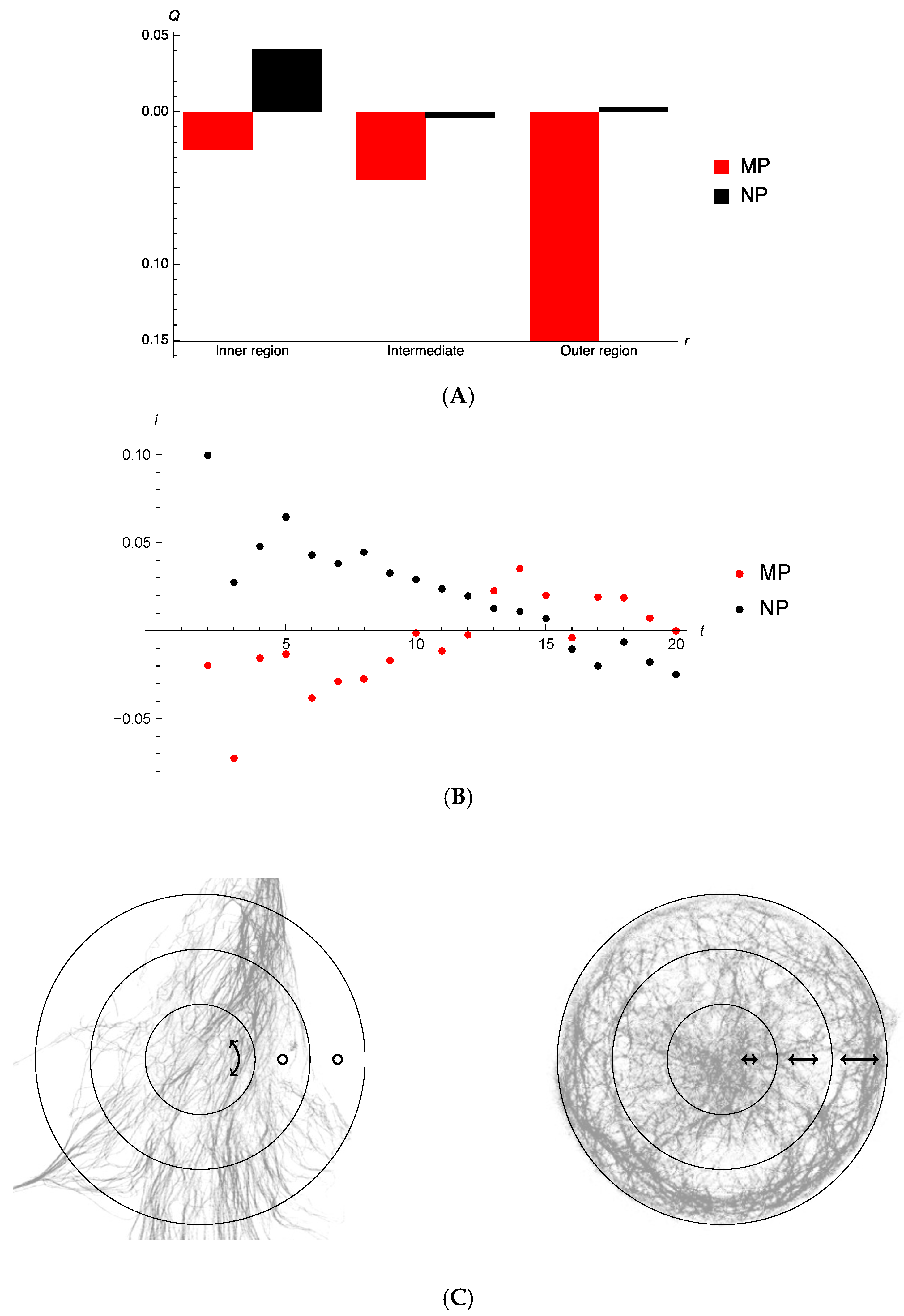{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Fabrication of Micropatterned Slides
2.3. Treatment Conditions
2.4. Live Cell Imaging
2.5. Image Analysis
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
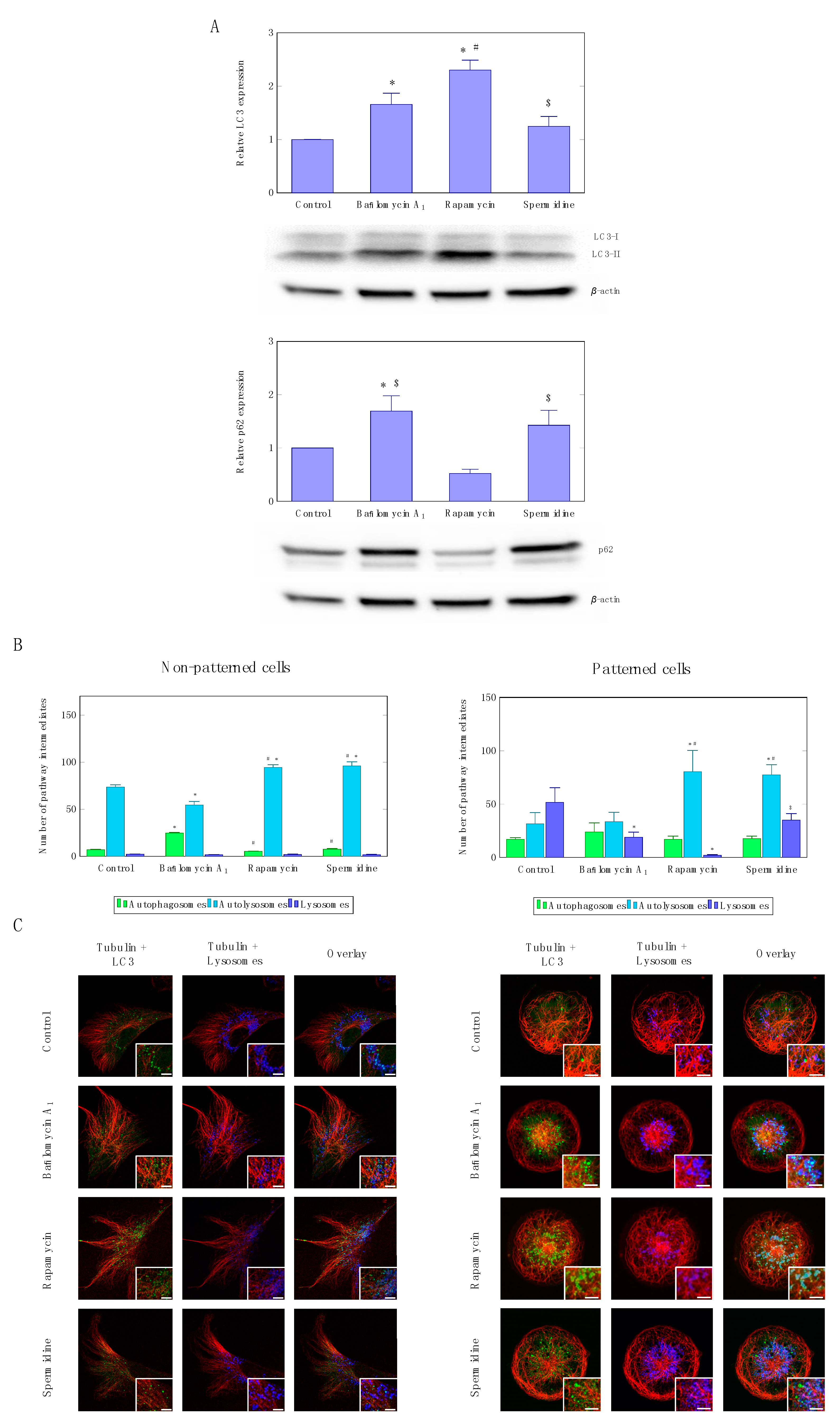
3.1. Precision Control of Autophagic Flux—Achieving Similar Autophagic Activity Via mTOR Dependent and Independent Pathway Induction
3.2. Precision Control of Cell Size and Tubulin Network—Assessment of Pool Sizes and Vesicle Trafficking
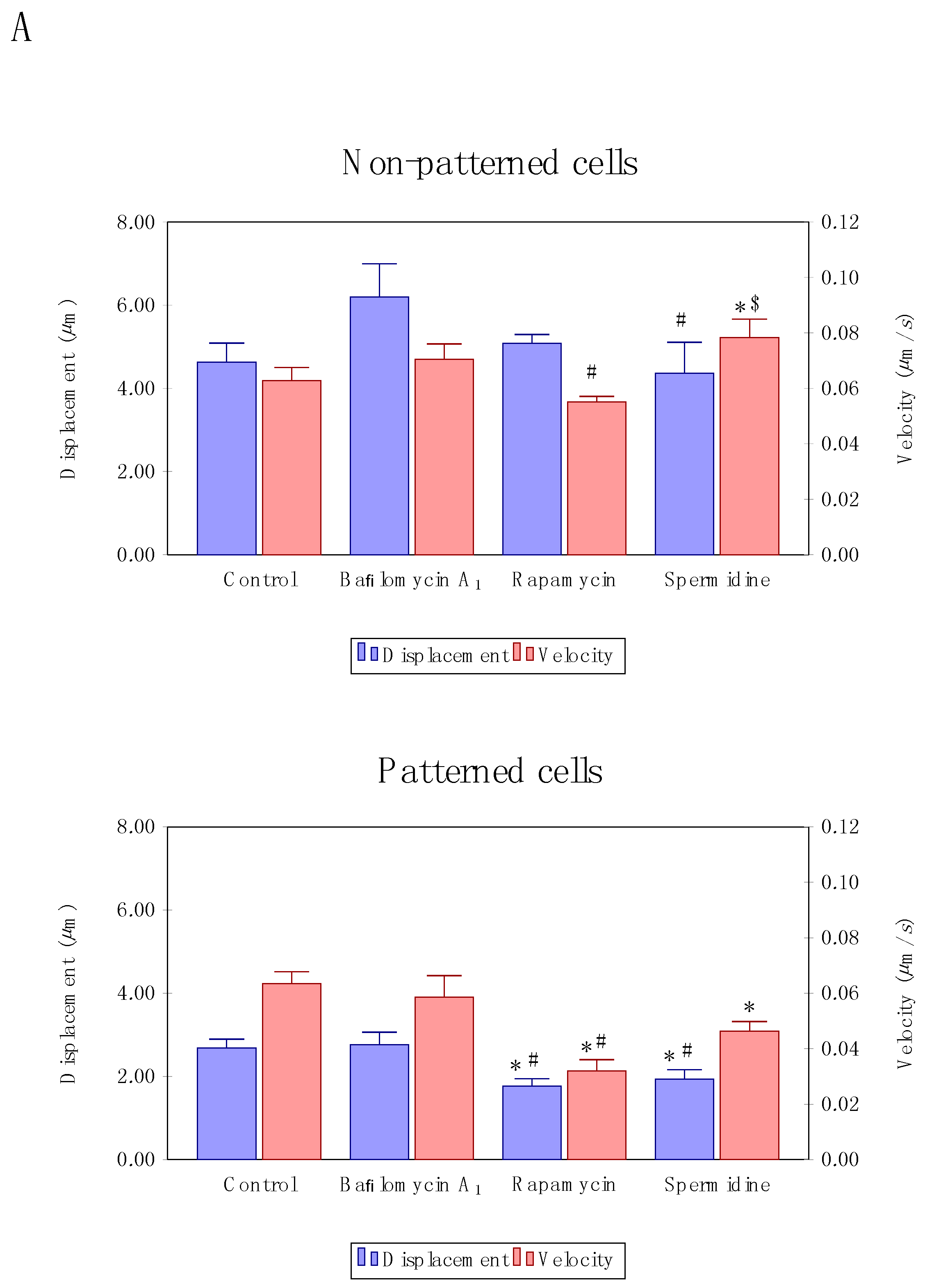
3.3. Precision Control of Cell Size and Tubulin Network—Assessment of Autophagosome Trafficking
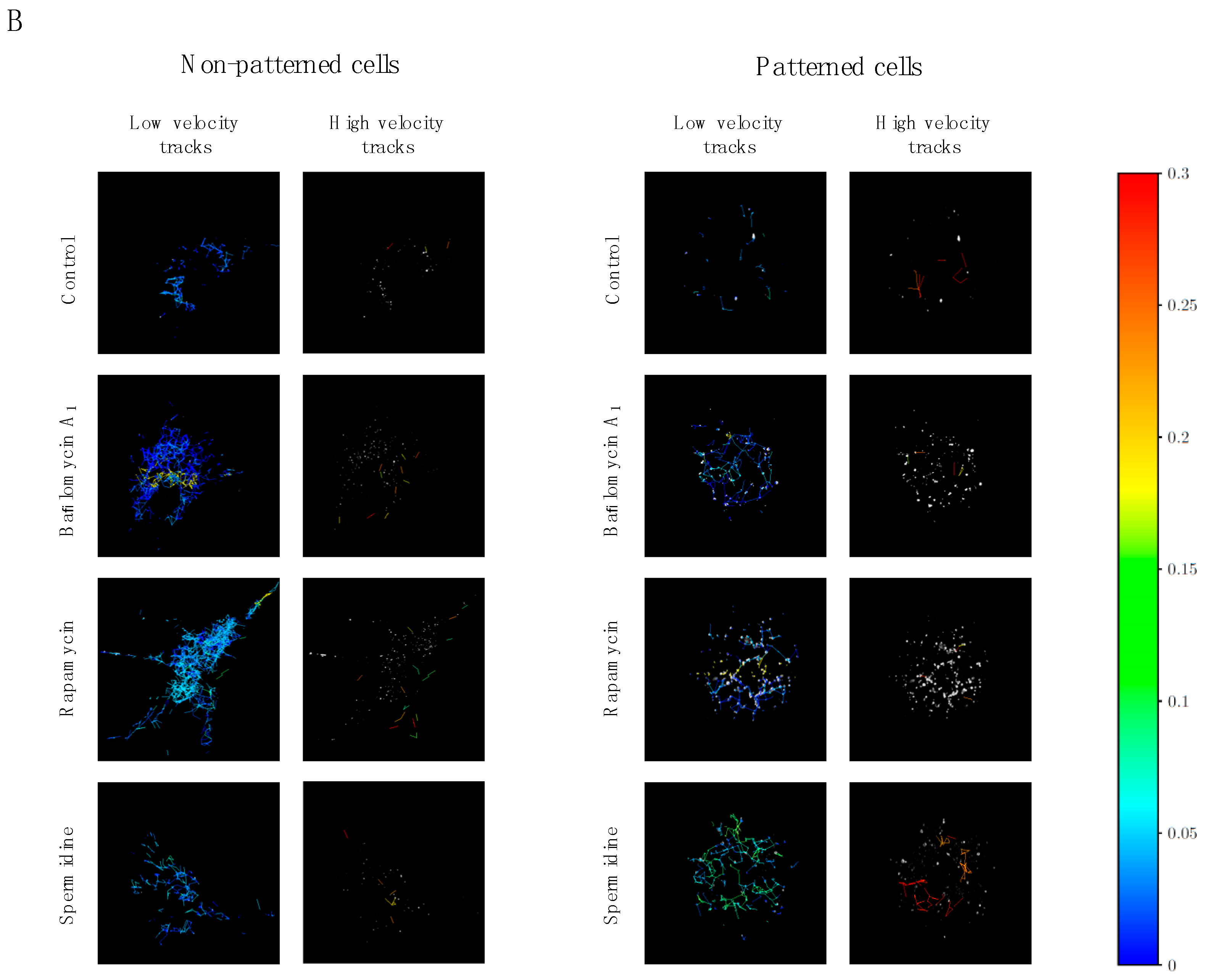
3.4. Directionality of Autophagosome Motion
4. Discussion
4.1. Autophagic Flux Control is Achievable
4.2. Enhanced Autophagic Flux Decreases Autophagosome Velocity
4.3. Cell Morphology and Tubulin Network Geometry Influence Autophagosome Motion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. In Seminars in Cell & Developmental Biology; Academic Press: New York, NY, USA, 2010; pp. 683–690. [Google Scholar]
- Loos, B.; Klionsky, D.J.; Wong, E. Augmenting brain metabolism to increase macro-and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Prog. Neurobiol. 2017, 156, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.; Engelbrecht, A.M. Cell death: A dynamic response concept. Autophagy 2009, 5, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.; Engelbrecht, A.M.; Lockshin, R.A.; Klionsky, D.J.; Zakeri, Z. The variability of autophagy and cell death susceptibility: Unanswered questions. Autophagy 2013, 9, 1270–1285. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Haspel, J.; Shaik, R.S.; Ifedigbo, E.; Nakahira, K.; Dolinay, T.; Englert, J.A.; Choi, A.M. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 2011, 7, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Ntsapi, C.; Loos, B. Caloric restriction and the precision-control of autophagy: A strategy for delaying neurodegenerative disease progression. Exp. Gerontol. 2016, 83, 97–111. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An autophagic flux probe that releases an internal control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.C.; Maziarz, R.T.; Spurgeon, S.E.; Medvedova, E.; Gajewski, J.; Reasor-Heard, S.; Park, B.; Kratz, A.; Thomas, G.V.; Loriaux, M.; et al. Double autophagy stimulation using chemotherapy and mTOR inhibition combined with hydroxychloroquine for autophagy modulation in patients with relapsed or refractory multiple myeloma. Haematologica 2017, 102, e261–e265. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.; Toit, A.D.; Hofmeyr, J.H.S. Defining and measuring autophagosome flux—Concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Peña-Oyarzun, D.; Troncoso, R.; Kretschmar, C.; Hernando, C.; Budini, M.; Morselli, E.; Lavandero, S.; Criollo, A. Hyperosmotic stress stimulates autophagy via polycystin-2. Oncotarget 2017, 8, 55984–55997. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J. Autophagy research: Lessons from metabolism. Autophagy 2009, 5, 3–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Du Toit, A.; Hofmeyr, J.S.H.; Gniadek, T.J.; Loos, B. Measuring Autophagosome Flux. Autophagy 2018. Accepted. [Google Scholar]
- Kane, M.S.; Alban, J.; Desquiret-Dumas, V.; Gueguen, N.; Ishak, L.; Ferre, M.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Lenaers, G.; et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J. Cell. Mol. Med. 2017, 21, 2284–2297. [Google Scholar] [CrossRef] [PubMed]
- Carpi, N.; Piel, M.; Azioune, A.; Fink, J. Micropatterning on glass with deep UV. Nat. Protoc. Exch. 2011. [Google Scholar] [CrossRef]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, A.; Hofmeyr, J.H.S.; Loos, B. Methods for Measuring Autophagosome Flux—Impact and Relevance. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Academic Press: New York, NY, USA, 2017; pp. 91–104. [Google Scholar]
- Han, K.; Kim, J.; Choi, M.Y. Quantitative indices of autophagy activity from minimal models. Theor. Biol. Med. Model. 2014, 11, 31. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Finkbeiner, S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 2013, 9, 586–592. [Google Scholar]
- Yuan, J. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef] [PubMed]
- Lumkwana, D.; du Toit, A.; Kinnear, C.; Loos, B. Autophagic flux control in neurodegeneration: Progress and precision targeting—Where do we stand? Prog. Neurobiol. 2017, 153, 64–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, S.; Zhang, Y.; Lin, X.; Song, Y.; Xue, Z.; Qian, H.; Wang, S.; Wan, G.; Zheng, X.; et al. Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. 2017, 8, e2738. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Monastyrska, I.; Rieter, E.; Klionsky, D.J.; Reggiori, F. Multiple roles of the cytoskeleton in autophagy. Biol. Rev. 2009, 84, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Köchl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Fujita, N.; Yoshimori, T. The late stages of autophagy: How does the end begin? Cell Death Differ. 2009, 16, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, J.; Mulder, B.M.; Koenderink, G.H. Alignment of nematic and bundled semiflexible polymers in cell-sized confinement. Soft Matter 2014, 10, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Azari, A.; Müller-Nedebock, K.K. Entropic competition in polymeric systems under geometrical confinement. EPL Europhys. Lett. 2015, 110, 68004. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du Toit, A.; De Wet, S.; Hofmeyr, J.-H.S.; Müller-Nedebock, K.K.; Loos, B. The Precision Control of Autophagic Flux and Vesicle Dynamics—A Micropattern Approach. Cells 2018, 7, 94. https://doi.org/10.3390/cells7080094
Du Toit A, De Wet S, Hofmeyr J-HS, Müller-Nedebock KK, Loos B. The Precision Control of Autophagic Flux and Vesicle Dynamics—A Micropattern Approach. Cells. 2018; 7(8):94. https://doi.org/10.3390/cells7080094
Chicago/Turabian StyleDu Toit, André, Sholto De Wet, Jan-Hendrik S. Hofmeyr, Kristian K. Müller-Nedebock, and Ben Loos. 2018. "The Precision Control of Autophagic Flux and Vesicle Dynamics—A Micropattern Approach" Cells 7, no. 8: 94. https://doi.org/10.3390/cells7080094
APA StyleDu Toit, A., De Wet, S., Hofmeyr, J.-H. S., Müller-Nedebock, K. K., & Loos, B. (2018). The Precision Control of Autophagic Flux and Vesicle Dynamics—A Micropattern Approach. Cells, 7(8), 94. https://doi.org/10.3390/cells7080094