
Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles

{kind=link}
{kind=link}
Abstract
1. Introduction
Interaction of Rabs, Autophagy and Intracellular Bacteria
2. Intracellular Bacterial Pathogens: Interplay with Rabs and Autophagy
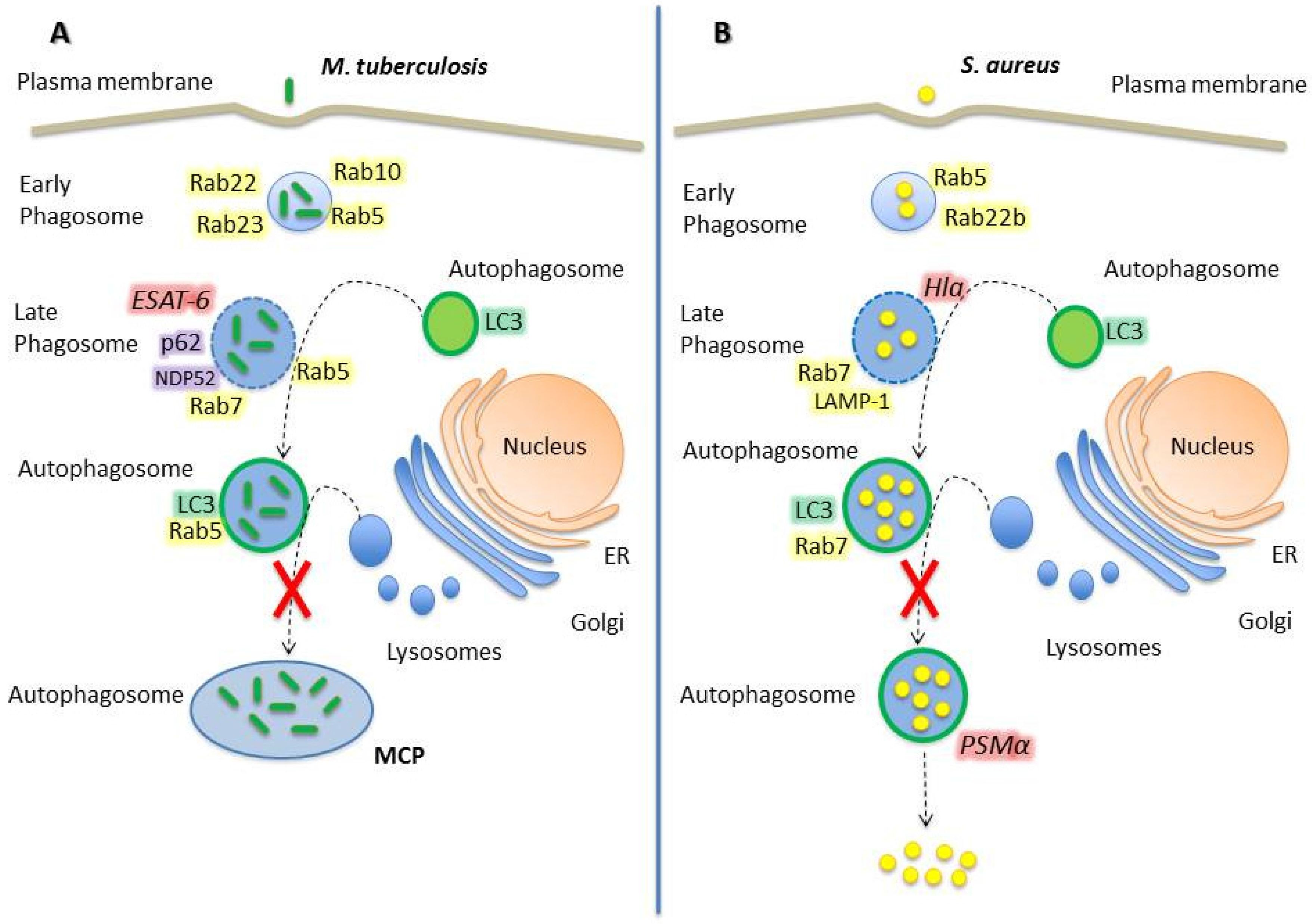
2.1. Mycobacterium Tuberculosis
2.2. Staphylococcus Aureus
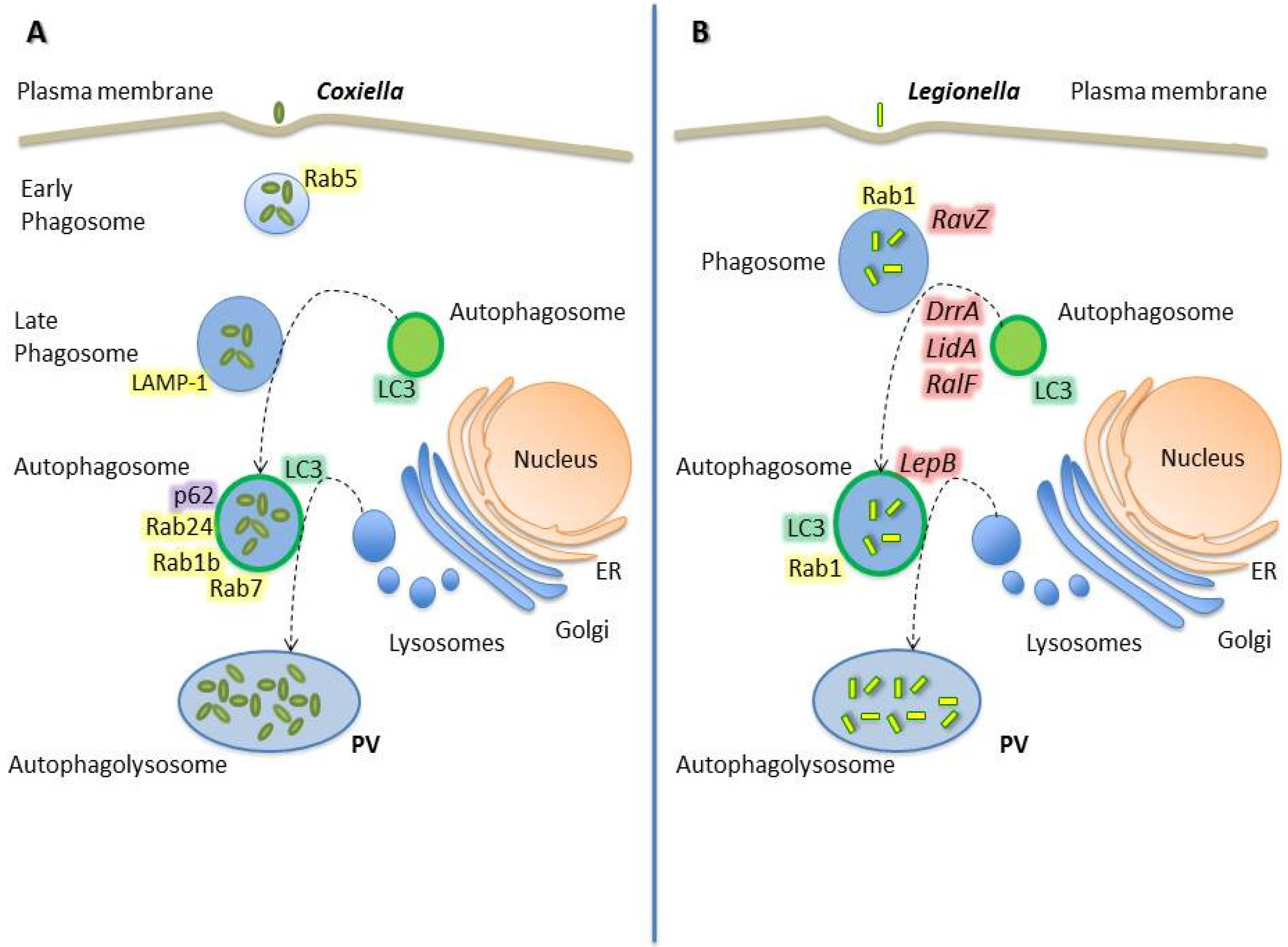
2.3. Coxiella Burnetii
2.4. Legionella Pneumophila
3. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stenmark, H.; Olkkonen, V.M. The Rab GTPase family. Genome Biol. 2001, 2, REVIEWS3007. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Chiariello, M. Signal transduction gRABs attention. Cell. Signal. 2006, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zerial, M.; Mcbride, H. Rab Proteins As Membrane Organizers. Nat. Rev. Mol. Cell Biol. 2001, 2. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leal, J.B.; Seabra, M.C. Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 2001, 313, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yin, G.; Zhao, X.; Ji, C.; Gu, S.; Tang, R.; Dong, H.; Xie, Y.; Mao, Y. Human RAB24, interestingly and predominantly distributed in the nuclei of COS-7 cells, is colocalized with cyclophilin A and GABARAP. Int. J. Mol. Med. 2006, 17, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.L.; Cao, C.; Pylypenko, O.; Rak, A.; Wandinger-Ness, A. Rab GTPases at a glance. J. Cell Sci. 2008, 121, 246–246. [Google Scholar] [CrossRef]
- Vonderheit, A.; Helenius, A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005, 3, e233. [Google Scholar] [CrossRef] [PubMed]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Hernández-González, J.C.; Luna-Herrera, J.; García-Pérez, B.E. The role of autophagy in bacterial infections. Biosci. Trends 2015, 9, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Sorbara, M.T.; Philpott, D.J.; Girardin, S.E. Bacterial autophagy. Autophagy 2012, 8, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Levine, B. Autophagy genes in immunity. Nat. Immunol. 2009, 10, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S. Autophagy and bacterial clearance: a not so clear picture. Cell. Microbiol. 2013, 15, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Méresse, S.; Steele-Mortimer, O.; Moreno, E.; Desjardins, M.; Finlay, B.; Gorvel, J.P. Controlling the maturation of pathogen-containing vacuoles: A matter of life and death. Nat. Cell Biol. 1999, 1, E183–E188. [Google Scholar] [PubMed]
- Vieira, O.V.; Botelho, R.J.; Grinstein, S. Phagosome maturation: Aging gracefully. Biochem. J. 2002, 366, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Duclos, S.; Desjardins, M. Subversion of a young phagosome: The survival strategies of intracellular pathogens. Cell. Microbiol. 2000, 2, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Löffler, B.; Tuchscherr, L.; Niemann, S.; Peters, G. Staphylococcus aureus persistence in non-professional phagocytes. Int. J. Med. Microbiol. 2014, 304, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Mwandumba, H.C.; Rhoades, E.E. Mycobacterium and the coat of many lipids. J. Cell Biol. 2002, 158, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Howe, D.; Shannon, J.G.; Winfree, S.; Dorward, D.W.; Heinzen, R.A. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect. Immun. 2010, 78, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- Prashar, A.; Terebiznik, M.R. Legionella pneumophila: homeward bound away from the phagosome. Curr. Opin. Microbiol. 2015, 23, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.M. Cell-mediated immune responses in tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 2012, 12, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Seto, S.; Tsujimura, K.; Koide, Y. Rab GTPases regulating phagosome maturation are differentially recruited to mycobacterial phagosomes. Traffic 2011, 12, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Chua, J.; Kyei, G.B.; Deretic, V. Higher order Rab programming in phagolysosome biogenesis. J. Cell Biol. 2006, 174, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Vieira, O.V.; Botelho, R.J.; Rameh, L.; Brachmann, S.M.; Matsuo, T.; Davidson, H.W.; Schreiber, A.; Backer, J.M.; Cantley, L.C.; Grinstein, S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 2001, 155, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Fratti, R.A.; Chua, J.; Vergne, I.; Deretic, V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A 2003, 100, 5437–5442. [Google Scholar] [CrossRef] [PubMed]
- Fratti, R.A.; Backer, J.M.; Gruenberg, J.; Corvera, S.; Deretic, V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol. 2001, 154, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Via, L.E.; Deretic, D.; Ulmer, R.J.; Hibler, N.S.; Huber, L.A.; Deretic, V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 1997, 272, 13326–13331. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.-P.; Müller, M.P.; Wandinger-Ness, A. Bacterial pathogens commandeer Rab GTPases to establish intracellular niches. Traffic 2012, 13, 1565–1588. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.M. P.; Jordao, L.; Vieira, O.V. Rab10 regulates phagosome maturation and its overexpression rescues Mycobacterium-containing phagosomes maturation. Traffic 2010, 11, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Simeone, R.; Bottai, D.; Frigui, W.; Majlessi, L.; Brosch, R. ESX/type VII secretion systems of mycobacteria: Insights into evolution, pathogenicity and protection. Tuberculosis (Edinb). 2015, 95, S150–S154. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Sun, J. Mechanism of ESAT-6 membrane interaction and its roles in pathogenesis of Mycobacterium tuberculosis. Toxicon 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Keil, V.; Sun, J. Characterization of Mycobacterium tuberculosis EsxA membrane insertion: Roles of N- and C-terminal flexible arms and central helix-turn-helix motif. J. Biol. Chem. 2015, 290, 7314–7322. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Beaumelle, B.; Vergne, I. Autophagy in Mycobacterium tuberculosis and HIV infections. Front. Cell. Infect. Microbiol. 2015, 5, 1–8. [Google Scholar]
- Chandra, P.; Ghanwat, S.; Matta, S.K.; Yadav, S.S.; Mehta, M.; Siddiqui, Z.; Singh, A.; Kumar, D. Mycobacterium tuberculosis Inhibits RAB7 Recruitment to Selectively Modulate Autophagy Flux in Macrophages. Sci. Rep. 2015, 5, 16320. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.F.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.A.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.S.; Bhattacharya, D.; et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. 2012, 109, 3168–3176. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.J.; Foster, T.J.; Cameron, B.J.; Berendt, A.R. Bacterial fibronectin-binding proteins and endothelial cell surface fibronectin mediate adherence of Staphylococcus aureus to resting human endothelial cells. Microbiology 1999, 145, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; François, P.P.; Nüsse, O.; Foti, M.; Hartford, O.M.; Vaudaux, P.; Foster, T.J.; Lew, D.P.; Herrmann, M.; Krause, K.H. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell. Microbiol. 1999, 1, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.; Wann, E.R.; Joh, D.; Johansson, S.; Foster, T.J.; Höök, M. Cellular invasion by Staphylococcus aureus involves a fibronectin bridge between the bacterial fibronectin-binding MSCRAMMs and host cell beta1 integrins. Eur. J. Cell Biol. 2000, 79, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Fraunholz, M. Staphylococcus aureus host cell invasion and post-invasion events. Int. J. Med. Microbiol. 2010, 300, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Jarry, T.M.; Cheung, A.L. Staphylococcus aureus escapes more efficiently from the phagosome of a cystic fibrosis bronchial epithelial cell line than from its normal counterpart. Infect. Immun. 2006, 74, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; et al. A potential new pathway for Staphylococcus aureus dissemination: The silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 2008, 3, e1409. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.L.; Wang, Y.; Tang, B.L. Rab22B’s role in trans-Golgi network membrane dynamics. Biochem. Biophys. Res. Commun. 2007, 361, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Kokubu, E.; Sugiura, S.; Komatsu, T.; Tada, H.; Isoda, R.; Tanigawa, N.; Kato, Y.; Ishida, N.; Kobayashi, K.; et al. Vinculin and Rab5 complex is requited for uptake of Staphyrococcus aureus and interleukin-6 expression. PLoS One 2014, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Del Nery, E.; Miserey-Lenkei, S.; Falguières, T.; Nizak, C.; Johannes, L.; Perez, F.; Goud, B. Rab6A and Rab6A’ GTPases play non-overlapping roles in membrane trafficking. Traffic 2006, 7, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Stauber, T.; del Nery, E.; Vernos, I.; Pepperkok, R.; Nilsson, T. Regulation of microtubule-dependent recycling at the trans-Golgi network by Rab6A and Rab6A’. Mol. Biol. Cell 2005, 16, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Opdam, F.J.; Echard, A.; Croes, H.J.; van den Hurk, J.A.; van de Vorstenbosch, R.A.; Ginsel, L.A.; Goud, B.; Fransen, J.A. The small GTPase Rab6B, a novel Rab6 subfamily member, is cell-type specifically expressed and localised to the Golgi apparatus. J. Cell Sci. 2000, 113, 2725–2735. [Google Scholar] [PubMed]
- Chen, Y.; Jiang, C.; Jin, M.; Gong, Y.; Zhang, X. The role of Rab6 GTPase in the maturation of phagosome against Staphylococcus aureus. Int. J. Biochem. Cell Biol. 2015, 61, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Jarry, T.M.; Memmi, G.; Cheung, A.L. The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell. Microbiol. 2008, 10, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Perskvist, N.; Roberg, K.; Kulyté, A.; Stendahl, O. Rab5a GTPase regulates fusion between pathogen-containing phagosomes and cytoplasmic organelles in human neutrophils. J. Cell Sci. 2002, 115, 1321–1330. [Google Scholar] [PubMed]
- Mestre, M.B.; Fader, C.M.; Sola, C.; Colombo, M.I. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy 2010, 6, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Kneuper, H.; Cao, Z.P.; Twomey, K.B.; Zoltner, M.; Jäger, F.; Cargill, J.S.; Chalmers, J.; van der Kooi-Pol, M.M.; van Dijl, J.M.; Ryan, R.P.; et al. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/Type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol. Microbiol. 2014, 93, 928–943. [Google Scholar] [CrossRef] [PubMed]
- Schnaith, A.; Kashkar, H.; Leggio, S.A.; Addicks, K.; Krönke, M.; Krut, O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 2007, 282, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.C.; Schäfer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell. Microbiol. 2014, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [PubMed]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mestre, M.B.; Colombo, M.I. cAMP and EPAC are key players in the regulation of the signal transduction pathway involved in the α-hemolysin autophagic response. PLoS Pathog. 2012, 8, e1002664. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.I.; Gutierrez, M.G.; Romano, P.S. The two faces of autophagy: Coxiella and Mycobacterium. Autophagy 2006, 2, 162–164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Voth, D.E.; Heinzen, R.A. Lounging in a lysosome: The intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 2007, 9, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Heinzen, R.A. Coxiella type IV secretion and cellular microbiology. Curr. Opin. Microbiol. 2009, 12, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, C.A.; McDonough, J.A.; Newton, H.J.; Roy, C.R. The role of Rab GTPases in the transport of vacuoles containing Legionella pneumophila and Coxiella burnetii. Biochem. Soc. Trans. 2012, 40, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Van Schaik, E.J.; Chen, C.; Mertens, K.; Weber, M.M.; Samuel, J.E. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat. Rev. Microbiol. 2013, 11, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.J.; Roy, C.R. Biogenesis of the lysosome-derived vacuole containing Coxiella burnetii. Microbes Infect. 17, 766–771. [CrossRef] [PubMed]
- Newton, H.J.; Kohler, L.J.; McDonough, J.A.; Temoche-Diaz, M.; Crabill, E.; Hartland, E.L.; Roy, C.R. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 2014, 10, e1004286. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.H.; Newton, P.; Newton, H.J. Coxiella burnetii: Turning hostility into a home. Cell. Microbiol. 2015, 17, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Brumell, J.H.; Scidmore, M. A Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol. Mol. Biol. Rev. 2007, 71, 636–652. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The role of membrane-trafficking small GTPases in the regulation of autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.S.; Gutierrez, M.G.; Berón, W.; Rabinovitch, M.; Colombo, M.I. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 2007, 9, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Berón, W.; Gutierrez, M.G.; Rabinovitch, M.; Colombo, M.I. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 2002, 70, 5816–5821. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Vázquez, C.L.; Munafó, D.B.; Zoppino, F.C.M.; Berón, W.; Rabinovitch, M.; Colombo, M.I. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell. Microbiol. 2005, 7, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Winchell, C.G.; Graham, J.G.; Kurten, R.C.; Voth, D.E. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect. Immun. 2014, 82, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Winchell, C.G.; Steele, S.; Kawula, T.; Voth, D.E. Dining in: Intracellular bacterial pathogen interplay with autophagy. Curr. Opin. Microbiol. 2015, 29, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Campoy, E.M.; Zoppino, F.C.M.; Colombo, M.I. The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect. Immun. 2011, 79, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.K.; Voth, D.E. Coxiella subversion of intracellular host signaling. Adv.Exp.Med.Biol. 2012, 984, 131–140. [Google Scholar] [PubMed]
- Vázquez, C.L.; Colombo, M.I. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2010, 17, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Finsel, I.; Otto, A.; Pfaffinger, G.; Rothmeier, E.; Hecker, M.; Becher, D.; Hilbi, H. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell. Microbiol. 2014, 16, 1034–1052. [Google Scholar] [PubMed]
- Hubber, A.; Roy, C.R. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 2010, 26, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Mousnier, A.; Schroeder, G.N.; Stoneham, C.A.; So, E.C.; Garnett, J.A; Yu, L.; Matthews, S.J.; Choudhary, J.S.; Hartland, E.L.; Frankel, G. A New Method To Determine In Vivo Interactomes Reveals Binding of the Legionella pneumophila Effector PieE to Multiple Rab GTPases. MBio 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Banga, S.; Gao, P.; Shen, X.; Fiscus, V.; Zong, W.-X.; Chen, L.; Luo, Z.-Q. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. USA. 2007, 104, 5121–5126. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Swanson, M.S. Secrets of a successful pathogen: Legionella resistance to progression along the autophagic pathway. Front. Microbiol. 2011, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amer, A.O.; Swanson, M.S. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell. Microbiol. 2005, 7, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Roy, C.R. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 2002, 4, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.-F.; Swanson, M.S. Mouse infection by Legionella, a model to analyze autophagy. Autophagy 2, 179–182. [CrossRef]
- Horenkamp, F.A.; Kauffman, K.J.; Kohler, L.J.; Sherwood, R.K.; Krueger, K.P.; Shteyn, V.; Roy, C.R.; Melia, T.J.; Reinisch, K.M. The Legionella Anti-autophagy Effector RavZ Targets the Autophagosome via PI3P- and Curvature-Sensing Motifs. Dev. Cell 2015, 34, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Sturgill-Koszycki, S.; Swanson, M.S. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 2000, 192, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Goody, R.S.; Itzen, A. Modulation of small GTPases by Legionella. Curr. Top. Microbiol. Immunol. 2013, 376, 117–133. [Google Scholar] [PubMed]
- Müller, M.P.; Peters, H.; Blümer, J.; Blankenfeldt, W.; Goody, R.S.; Itzen, A. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 2010, 329, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Mihai Gazdag, E.; Streller, A.; Haneburger, I.; Hilbi, H.; Vetter, I.R.; Goody, R.S.; Itzen, A. Mechanism of Rab1b deactivation by the Legionella pneumophila GAP LepB. EMBO Rep. 2013, 14, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Neunuebel, M.R.; Machner, M.P. The taming of a Rab GTPase by Legionella pneumophila. Small GTPases 2012, 3, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tascón, I.; Neunuebel, M.R.; Pallara, C.; Brady, J.; Kinch, L.N.; Fernández-Recio, J.; Rojas, A.L.; Machner, M.P.; Hierro, A. Structural basis for Rab1 de-AMPylation by the Legionella pneumophila effector SidD. PLoS Pathog. 2013, 9, e1003382. [Google Scholar] [CrossRef]
- Sohn, Y.-S.; Shin, H.-C.; Park, W.S.; Ge, J.; Kim, C.-H.; Lee, B.L.; Heo, W. Do; Jung, J.U.; Rigden, D.J.; Oh, B.-H. Lpg0393 of Legionella pneumophila is a guanine-nucleotide exchange factor for Rab5, Rab21 and Rab22. PLoS One 2015, 10, e0118683. [Google Scholar] [CrossRef] [PubMed]
- Neunuebel, M.R.; Mohammadi, S.; Jarnik, M.; Machner, M.P. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J. Bacteriol. 2012, 194, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, E.; Pfaffinger, G.; Hoffmann, C.; Harrison, C.F.; Grabmayr, H.; Repnik, U.; Hannemann, M.; Wölke, S.; Bausch, A.; Griffiths, G.; et al. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog. 2013, 9, e1003598. [Google Scholar] [CrossRef] [PubMed]
- Manske, C.; Hilbi, H. Metabolism of the vacuolar pathogen Legionella and implications for virulence. Front. Cell. Infect. Microbiol. 2014, 4, 125. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López de Armentia, M.M.; Amaya, C.; Colombo, M.I. Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles. Cells 2016, 5, 11. https://doi.org/10.3390/cells5010011
López de Armentia MM, Amaya C, Colombo MI. Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles. Cells. 2016; 5(1):11. https://doi.org/10.3390/cells5010011
Chicago/Turabian StyleLópez de Armentia, María Milagros, Celina Amaya, and María Isabel Colombo. 2016. "Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles" Cells 5, no. 1: 11. https://doi.org/10.3390/cells5010011
APA StyleLópez de Armentia, M. M., Amaya, C., & Colombo, M. I. (2016). Rab GTPases and the Autophagy Pathway: Bacterial Targets for a Suitable Biogenesis and Trafficking of Their Own Vacuoles. Cells, 5(1), 11. https://doi.org/10.3390/cells5010011