
Breaking the Feedback Loop of β-Cell Failure: Insight into the Pancreatic β-Cell’s ER-Mitochondria Redox Balance


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
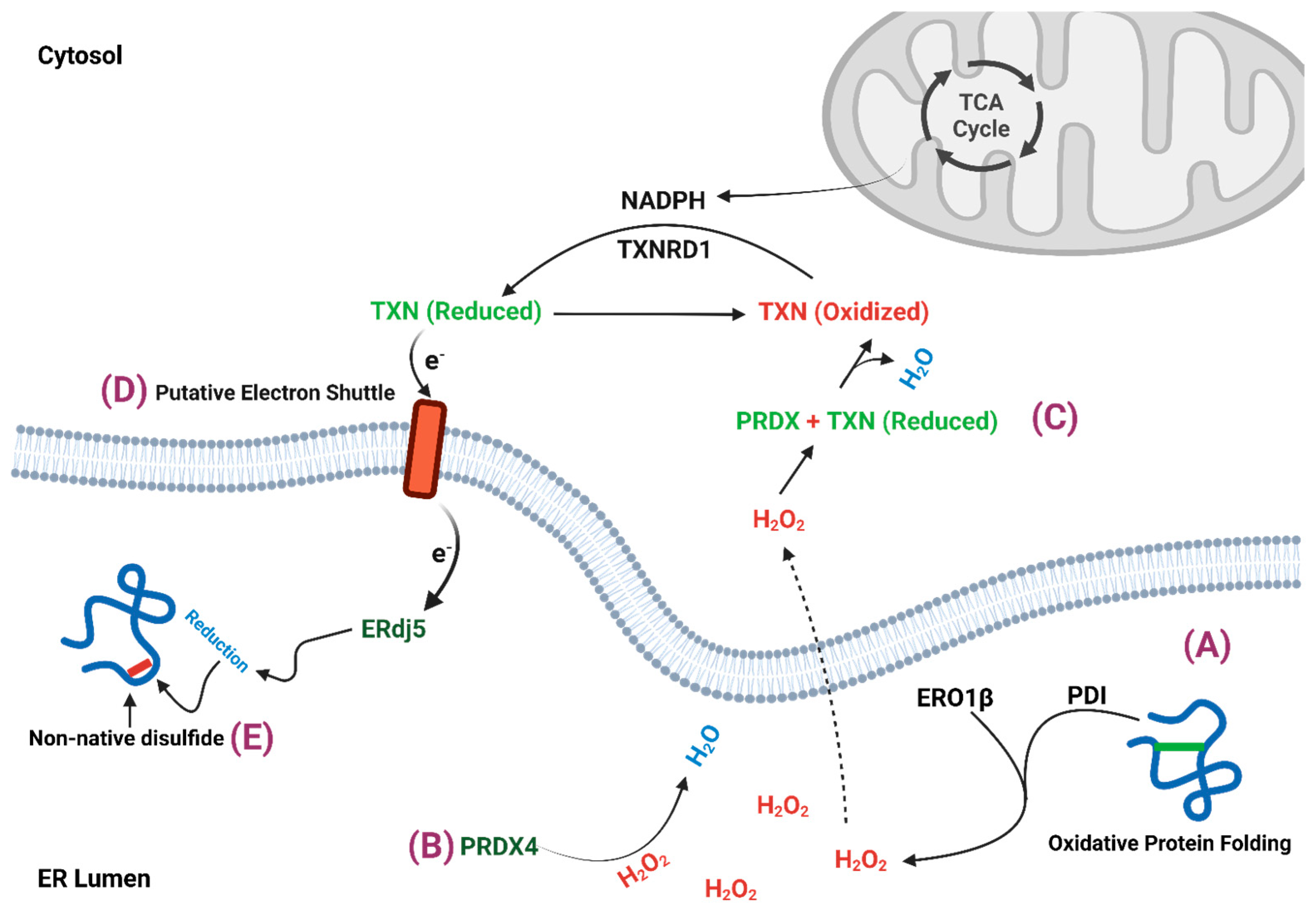
2. β-Cell ER Redox Control
2.1. ERO1
2.2. Peroxiredoxin 4
2.3. Thioredoxin
3. Mitochondrial Regulation of ER Redox Homeostasis
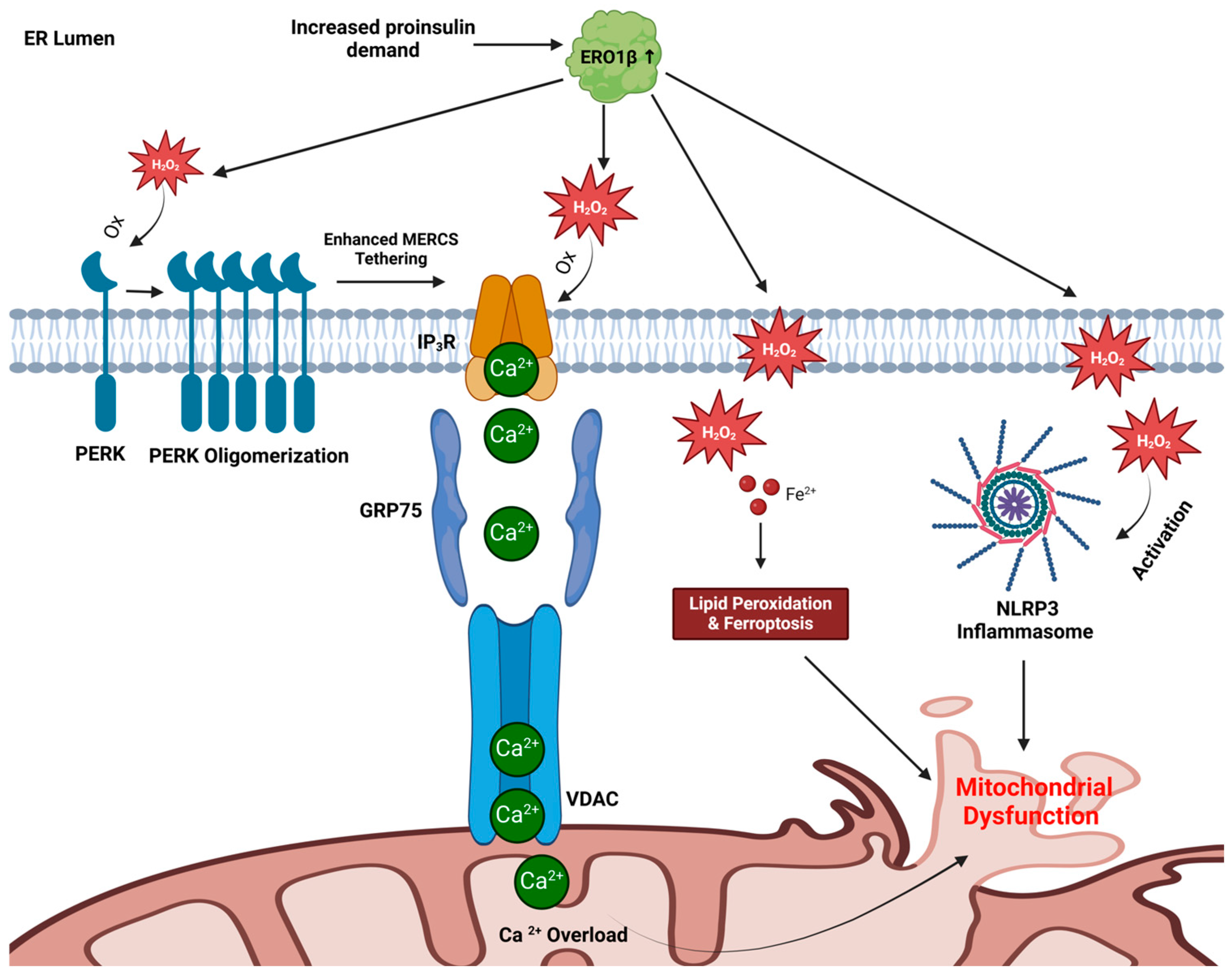
4. Trading Places: Can ER Redox Alter Mitochondrial Function in β-Cells?
4.1. Mitochondrial Calcium Overload
4.2. Ferroptosis Induction
4.3. NLRP3 Inflammasome Activation
4.4. A Model for ER-Directed Mitochondrial Dysfunction
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rohli, K.E.; Boyer, C.K.; Blom, S.E.; Stephens, S.B. Nutrient Regulation of Pancreatic Islet β-Cell Secretory Capacity and Insulin Production. Biomolecules 2022, 12, 335. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. J. Clin. Endocrinol. Metab. 2014, 99, 1983–1992. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Schuit, F.C.; Kiekens, R.; Pipeleers, D.G. Measuring the balance between insulin synthesis and insulin release. Biochem. Biophys. Res. Commun. 1991, 178, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Schuit, F.C.; In’t Veld, P.A.; Pipeleers, D.G. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3865–3869. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.B.; Rhodes, C.J.; Grimsby, J.S. The dynamic plasticity of insulin production in β-cells. Mol. Metab. 2017, 6, 958–973. [Google Scholar] [CrossRef]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20 (Suppl. S2), 28–50. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Klappa, P.; Freedman, R.B.; Lilie, H.; Rudolph, R. Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem. 2002, 277, 310–317. [Google Scholar] [CrossRef]
- Liu, M.; Li, Y.; Cavener, D.; Arvan, P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 13209–13212. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Arunagiri, A.; Hassan, A.; Regan, K.; Tsai, B.; Dhayalan, B.; Weiss, M.A.; Liu, M.; Arvan, P. Distinct states of proinsulin misfolding in MIDY. Cell. Mol. Life Sci. 2021, 78, 6017–6031. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Torres, M.; Zhang, J.; Lu, Y.; Haataja, L.; Reinert, R.B.; Knupp, J.; Chen, Y.J.; Parlakgul, G.; Arruda, A.P.; et al. Integration of ER protein quality control mechanisms defines β cell function and ER architecture. J. Clin. Investig. 2023, 133, e163584. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Frankær, C.G.; Knudsen, M.V.; Norén, K.; Nazarenko, E.; Ståhl, K.; Harris, P. The structures of T6, T3R3 and R6 bovine insulin: Combining X-ray diffraction and absorption spectroscopy. Acta Crystallogr. D Biol. Crystallogr. 2012, 68 Pt 10, 1259–1271. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–w437. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef]
- Zaher, A.; Petronek, M.S.; Allen, B.G.; Mapuskar, K.A. Balanced Duality: H2O2-Based Therapy in Cancer and Its Protective Effects on Non-Malignant Tissues. Int. J. Mol. Sci. 2024, 25, 8885. [Google Scholar] [CrossRef]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflug. Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef]
- Stancill, J.S.; Corbett, J.A. The Role of Thioredoxin/Peroxiredoxin in the β-Cell Defense Against Oxidative Damage. Front. Endocrinol. 2021, 12, 718235. [Google Scholar] [CrossRef] [PubMed]
- Spégel, P.; Sharoyko, V.V.; Goehring, I.; Danielsson, A.P.; Malmgren, S.; Nagorny, C.L.; Andersson, L.E.; Koeck, T.; Sharp, G.W.; Straub, S.G.; et al. Time-resolved metabolomics analysis of β-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem. J. 2013, 450, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Rohli, K.E.; Stubbe, N.J.; Walker, E.M.; Pearson, G.L.; Soleimanpour, S.A.; Stephens, S.B. A metabolic redox relay supports ER proinsulin export in pancreatic islet β cells. JCI Insight 2024, 9, e178725. [Google Scholar] [CrossRef]
- Rohli, K.E.; Boyer, C.K.; Bearrows, S.C.; Moyer, M.R.; Elison, W.S.; Bauchle, C.J.; Blom, S.E.; Zhang, J.; Wang, Y.; Stephens, S.B. ER Redox Homeostasis Regulates Proinsulin Trafficking and Insulin Granule Formation in the Pancreatic Islet β-Cell. Function 2022, 3, zqac051. [Google Scholar] [CrossRef] [PubMed]
- Arunagiri, A.; Haataja, L.; Cunningham, C.N.; Shrestha, N.; Tsai, B.; Qi, L.; Liu, M.; Arvan, P. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann. N. Y. Acad. Sci. 2018, 1418, 5–19. [Google Scholar] [CrossRef]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Frand, A.R.; Kaiser, C.A. The ERO1 Gene of Yeast Is Required for Oxidation of Protein Dithiols in the Endoplasmic Reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. The FAD- and O2-Dependent Reaction Cycle of Ero1-Mediated Oxidative Protein Folding in the Endoplasmic Reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Sevier, C.S.; Qu, H.; Heldman, N.; Gross, E.; Fass, D.; Kaiser, C.A. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 2007, 129, 333–344. [Google Scholar] [CrossRef]
- Zito, E.; Chin, K.-T.; Blais, J.; Harding, H.P.; Ron, D. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 2010, 188, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Khoo, C.; Yang, J.; Rajpal, G.; Wang, Y.; Liu, J.; Arvan, P.; Stoffers, D.A. Endoplasmic reticulum oxidoreductin-1-like β (ERO1lβ) regulates susceptibility to endoplasmic reticulum stress and is induced by insulin flux in β-cells. Endocrinology 2011, 152, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.G.; Søltoft, C.L.; Schmidt, J.D.; Birk, J.; Appenzeller-Herzog, C.; Ellgaard, L. Biochemical evidence that regulation of Ero1β activity in human cells does not involve the isoform-specific cysteine 262. Biosci. Rep. 2014, 34, e00103. [Google Scholar] [CrossRef]
- Enyedi, B.; Várnai, P.; Geiszt, M. Redox State of the Endoplasmic Reticulum Is Controlled by Ero1L-alpha and Intraluminal Calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Awazawa, M.; Futami, T.; Sakada, M.; Kaneko, K.; Ohsugi, M.; Nakaya, K.; Terai, A.; Suzuki, R.; Koike, M.; Uchiyama, Y.; et al. Deregulation of pancreas-specific oxidoreductin ERO1β in the pathogenesis of diabetes mellitus. Mol. Cell Biol. 2014, 34, 1290–1299. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 8, e44532. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Pagani, M.; Fabbri, M.; Benedetti, C.; Fassio, A.; Pilati, S.; Bulleid, N.J.; Cabibbo, A.; Sitia, R. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 2000, 275, 23685–23692. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Avezov, E.; Jörns, A.; Lenzen, S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E β-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radic. Biol. Med. 2017, 112, 121–130. [Google Scholar] [CrossRef]
- Tavender, T.J.; Bulleid, N.J. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 2010, 123 Pt 15, 2672–2679. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. Embo. J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Karplus, P.A.; Poole, L.B. Typical 2-Cys peroxiredoxins—Structures, mechanisms and functions. FEBS J. 2009, 276, 2469–2477. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Pinho Melo, E.; Lopes, C.; Mehmeti, I.; Lenzen, S.; Ron, D.; Avezov, E. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J. Cell Biol. 2015, 211, 253–259. [Google Scholar] [CrossRef]
- Ding, Y.; Yamada, S.; Wang, K.-Y.; Shimajiri, S.; Guo, X.; Tanimoto, A.; Murata, Y.; Kitajima, S.; Watanabe, T.; Izumi, H.; et al. Overexpression of Peroxiredoxin 4 Protects Against High-Dose Streptozotocin-Induced Diabetes by Suppressing Oxidative Stress and Cytokines in Transgenic Mice. Antioxid. Redox Signal. 2010, 13, 1477–1490. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 improves insulin biosynthesis and glucose-induced insulin secretion in insulin-secreting INS-1E cells. J. Biol. Chem. 2014, 289, 26904–26913. [Google Scholar] [CrossRef]
- Tran, D.T.; Pottekat, A.; Mir, S.A.; Loguercio, S.; Jang, I.; Campos, A.R.; Scully, K.M.; Lahmy, R.; Liu, M.; Arvan, P.; et al. Unbiased Profiling of the Human Proinsulin Biosynthetic Interaction Network Reveals a Role for Peroxiredoxin 4 in Proinsulin Folding. Diabetes 2020, 69, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Roscoe, J.M.; Sevier, C.S. Pathways for Sensing and Responding to Hydrogen Peroxide at the Endoplasmic Reticulum. Cells 2020, 9, 2314. [Google Scholar] [CrossRef]
- Elko, E.A.; Manuel, A.M.; White, S.; Zito, E.; van der Vliet, A.; Anathy, V.; Janssen-Heininger, Y.M.W. Oxidation of peroxiredoxin-4 induces oligomerization and promotes interaction with proteins governing protein folding and endoplasmic reticulum stress. J. Biol. Chem. 2021, 296, 100665. [Google Scholar] [CrossRef]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends. Biochem. Sci 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends. Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef]
- Gansemer, E.R.; McCommis, K.S.; Martino, M.; King-McAlpin, A.Q.; Potthoff, M.J.; Finck, B.N.; Taylor, E.B.; Rutkowski, D.T. NADPH and Glutathione Redox Link TCA Cycle Activity to Endoplasmic Reticulum Homeostasis. iScience 2020, 23, 101116. [Google Scholar] [CrossRef]
- Poet, G.J.; Oka, O.B.; van Lith, M.; Cao, Z.; Robinson, P.J.; Pringle, M.A.; Arnér, E.S.; Bulleid, N.J. Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. Embo J. 2017, 36, 693–702. [Google Scholar] [CrossRef]
- Kim, J.; Moon, J.Y.; Kim, W.J.; Kim, D.G.; Nam, B.H.; Kim, Y.O.; Park, J.Y.; An, C.M.; Kong, H.J. Molecular and Functional Characterization of Thioredoxin 1 from Korean Rose Bitterling (Rhodeus uyekii). Int. J. Mol. Sci. 2015, 16, 19433–19446. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- Chou, F.C.; Sytwu, H.K. Overexpression of thioredoxin in islets transduced by a lentiviral vector prolongs graft survival in autoimmune diabetic NOD mice. J. Biomed. Sci. 2009, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kutner, M.; Khomsky, L.; Trus, M.; Aisner, Y.; Niv, M.Y.; Benhar, M.; Atlas, D. Thioredoxin-mimetic peptides (TXM) reverse auranofin induced apoptosis and restore insulin secretion in insulinoma cells. Biochem. Pharmacol. 2013, 85, 977–990. [Google Scholar] [CrossRef]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. Faseb. J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A. Lack of TXNIP protects beta-cells against glucotoxicity. Biochem. Soc. Trans. 2008, 36 Pt 5, 963–965. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Thielen, L.A.; Chen, J.; Jing, G.; Moukha-Chafiq, O.; Xu, G.; Jo, S.; Grayson, T.B.; Lu, B.; Li, P.; Augelli-Szafran, C.E.; et al. Identification of an Anti-diabetic, Orally Available Small Molecule that Regulates TXNIP Expression and Glucagon Action. Cell Metab. 2020, 32, 353–365.e358. [Google Scholar] [CrossRef]
- Stancill, J.S.; Hansen, P.A.; Mathison, A.J.; Schmidt, E.E.; Corbett, J.A. Deletion of Thioredoxin Reductase Disrupts Redox Homeostasis and Impairs β-Cell Function. Function 2022, 3, zqac034. [Google Scholar] [CrossRef]
- Kadokura, H.; Katzen, F.; Beckwith, J. Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 2003, 72, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Lilla, S.; Cao, Z.; Pringle, M.A.; Oka, O.B.V.; Robinson, P.J.; Szmaja, T.; van Lith, M.; Zanivan, S.; Bulleid, N.J. The mammalian cytosolic thioredoxin reductase pathway acts via a membrane protein to reduce ER-localised proteins. J. Cell Sci. 2020, 133, jcs241976. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, R.; Quintens, R.; Dejonghe, S.; Tsukamoto, K.; in ’t Veld, P.; Renström, E.; Schuit, F.C. Redox control of exocytosis: Regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 2005, 54, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, T.M.; Ivarsson, R.; Li, D.Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renström, E. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 2009, 23, 893–900. [Google Scholar] [CrossRef]
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Investig. 2015, 125, 3847–3860. [Google Scholar] [CrossRef]
- Ronnebaum, S.M.; Ilkayeva, O.; Burgess, S.C.; Joseph, J.W.; Lu, D.; Stevens, R.D.; Becker, T.C.; Sherry, A.D.; Newgard, C.B.; Jensen, M.V. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 30593–30602. [Google Scholar] [CrossRef]
- Bauchle, C.J.; Rohli, K.E.; Boyer, C.K.; Pal, V.; Rocheleau, J.V.; Liu, S.; Imai, Y.; Taylor, E.B.; Stephens, S.B. Mitochondrial Efflux of Citrate and Isocitrate Is Fully Dispensable for Glucose-Stimulated Insulin Secretion and Pancreatic Islet β-Cell Function. Diabetes 2021, 70, 1717–1728. [Google Scholar] [CrossRef]
- Zhang, G.F.; Jensen, M.V.; Gray, S.M.; El, K.; Wang, Y.; Lu, D.; Becker, T.C.; Campbell, J.E.; Newgard, C.B. Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metab. 2021, 33, 804–817.e805. [Google Scholar] [CrossRef]
- Deepa Maheshvare, M.; Raha, S.; König, M.; Pal, D. A pathway model of glucose-stimulated insulin secretion in the pancreatic β-cell. Front. Endocrinol. 2023, 14, 1185656. [Google Scholar] [CrossRef] [PubMed]
- Pelligra, A.; Mrugala, J.; Griess, K.; Kirschner, P.; Nortmann, O.; Bartosinska, B.; Köster, A.; Krupenko, N.I.; Gebel, D.; Westhoff, P.; et al. Pancreatic islet protection at the expense of secretory function involves serine-linked mitochondrial one-carbon metabolism. Cell Rep. 2023, 42, 112703. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Jensen, M.V.; Hohmeier, H.E.; Burgess, S.C.; Zhou, Y.P.; Qian, S.; MacNeil, D.; Howard, A.; Thornberry, N.; Ilkayeva, O.; et al. Silencing of cytosolic or mitochondrial isoforms of malic enzyme has no effect on glucose-stimulated insulin secretion from rodent islets. J. Biol. Chem. 2008, 283, 28909–28917. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; Rutter, G.A. When less is more: The forbidden fruits of gene repression in the adult β-cell. Diabetes Obes. Metab. 2013, 15, 503–512. [Google Scholar] [CrossRef]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Odili, S.; Zelent, D.; Buettger, C.; Castleden, H.A.; Steele, A.M.; Stride, A.; Shiota, C.; Magnuson, M.A.; Lorini, R.; et al. Insights into the structure and regulation of glucokinase from a novel mutation (V62M), which causes maturity-onset diabetes of the young. J. Biol. Chem. 2005, 280, 14105–14113. [Google Scholar] [CrossRef]
- Martin, S.K.; Carroll, R.; Benig, M.; Steiner, D.F. Regulation by glucose of the biosynthesis of PC2, PC3 and proinsulin in (ob/ob) mouse islets of Langerhans. FEBS Lett. 1994, 356, 279–282. [Google Scholar] [CrossRef]
- Alarcón, C.; Lincoln, B.; Rhodes, C.J. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J. Biol. Chem. 1993, 268, 4276–4280. [Google Scholar] [CrossRef]
- Uchizono, Y.; Alarcón, C.; Wicksteed, B.L.; Marsh, B.J.; Rhodes, C.J. The balance between proinsulin biosynthesis and insulin secretion: Where can imbalance lead? Diabetes Obes. Metab. 2007, 9 (Suppl. S2), 56–66. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Wilson, E.L.; Metzakopian, E. ER-mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 2021, 28, 1804–1821. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ushioda, R.; Nagata, K. Redox states in the endoplasmic reticulum directly regulate the activity of calcium channel, inositol 1,4,5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2023, 120, e2216857120. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium. 2021, 94, 102356. [Google Scholar] [CrossRef]
- Bassot, A.; Chen, J.; Takahashi-Yamashiro, K.; Yap, M.C.; Gibhardt, C.S.; Le, G.N.T.; Hario, S.; Nasu, Y.; Moore, J.; Gutiérrez, T.; et al. The endoplasmic reticulum kinase PERK interacts with the oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep. 2023, 42, 111899. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Kennedy, E.D.; Rizzuto, R.; Theler, J.M.; Pralong, W.F.; Bastianutto, C.; Pozzan, T.; Wollheim, C.B. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J. Clin. Investig. 1996, 98, 2524–2538. [Google Scholar] [CrossRef]
- Walkon, L.L.; Strubbe-Rivera, J.O.; Bazil, J.N. Calcium Overload and Mitochondrial Metabolism. Biomolecules 2022, 12, 1891. [Google Scholar] [CrossRef]
- Madec, A.M.; Perrier, J.; Panthu, B.; Dingreville, F. Role of mitochondria-associated endoplasmic reticulum membrane (MAMs) interactions and calcium exchange in the development of type 2 diabetes. Int. Rev. Cell Mol. Biol. 2021, 363, 169–202. [Google Scholar] [CrossRef]
- Dingreville, F.; Panthu, B.; Thivolet, C.; Ducreux, S.; Gouriou, Y.; Pesenti, S.; Chauvin, M.-A.; Chikh, K.; Errazuriz-Cerda, E.; Van Coppenolle, F.; et al. Differential Effect of Glucose on ER-Mitochondria Ca2+ Exchange Participates in Insulin Secretion and Glucotoxicity-Mediated Dysfunction of β-Cells. Diabetes 2019, 68, 1778–1794. [Google Scholar] [CrossRef]
- Shu, J.; Gambardella, J.; Sorriento, D.; Santulli, G. Mechanistic Role of IP3R Calcium Release Channel in Pancreatic Beta-Cell Function. Diabetes 2018, 67 (Suppl. S1), 313-LB. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, L.; Guo, Z.; Wang, G.; Xu, J.; Zheng, Z.; Sun, K.; Guo, F. Lipid peroxidation in osteoarthritis: Focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov. 2023, 9, 320. [Google Scholar] [CrossRef]
- Nagasaki, T.; Schuyler, A.J.; Zhao, J.; Samovich, S.N.; Yamada, K.; Deng, Y.; Ginebaugh, S.P.; Christenson, S.A.; Woodruff, P.G.; Fahy, J.V.; et al. 15LO1 dictates glutathione redox changes in asthmatic airway epithelium to worsen type 2 inflammation. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Hansen, J.B.; Dos Santos, L.R.B.; Liu, Y.; Prentice, K.J.; Teudt, F.; Tonnesen, M.; Jonas, J.C.; Wheeler, M.B.; Mandrup-Poulsen, T. Glucolipotoxic conditions induce β-cell iron import, cytosolic ROS formation and apoptosis. J. Mol. Endocrinol. 2018, 61, 69–77. [Google Scholar] [CrossRef]
- Stancic, A.; Saksida, T.; Markelic, M.; Vucetic, M.; Grigorov, I.; Martinovic, V.; Gajic, D.; Ivanovic, A.; Velickovic, K.; Savic, N.; et al. Ferroptosis as a Novel Determinant of β-Cell Death in Diabetic Conditions. Oxid. Med. Cell Longev. 2022, 2022, 3873420. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Pepper, A.R.; Pawlick, R.L.; Gala-Lopez, B.; Gamble, A.F.; Kin, T.; Seeberger, K.; Korbutt, G.S.; Bornstein, S.R.; Linkermann, A.; et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Dixit, V.D. Nlrp3 Inflammasome Activation in Type 2 Diabetes: Is It Clinically Relevant? Diabetes 2012, 62, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Makio, T.; Simmen, T. Not So Rare: Diseases Based on Mutant Proteins Controlling Endoplasmic Reticulum-Mitochondria Contact (MERC) Tethering. Contact 2024, 7, 25152564241261228. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Kang, J.; He, Z.; Liu, Q.; Wu, J.; Li, D.; Wang, X.; Tao, Z.; Guan, X.; et al. Role of ROS-Induced NLRP3 Inflammasome Activation in the Formation of Calcium Oxalate Nephrolithiasis. Front. Immunol. 2022, 13, 818625. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, D.; Li, L.; Han, Y.; Dong, X.; Yang, L.; Li, X.; Li, W.; Li, W. Ginsenoside Rg1 ameliorates aging-induced liver fibrosis by inhibiting the NOX4/NLRP3 inflammasome in SAMP8 mice. Mol. Med. Rep. 2021, 24, 801. [Google Scholar] [CrossRef]
- Li, F.; Guan, Z.; Gao, Y.; Bai, Y.; Zhan, X.; Ji, X.; Xu, J.; Zhou, H.; Rao, Z. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol. Commun. 2024, 8, e0399. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaher, A.; Stephens, S.B. Breaking the Feedback Loop of β-Cell Failure: Insight into the Pancreatic β-Cell’s ER-Mitochondria Redox Balance. Cells 2025, 14, 399. https://doi.org/10.3390/cells14060399
Zaher A, Stephens SB. Breaking the Feedback Loop of β-Cell Failure: Insight into the Pancreatic β-Cell’s ER-Mitochondria Redox Balance. Cells. 2025; 14(6):399. https://doi.org/10.3390/cells14060399
Chicago/Turabian StyleZaher, Amira, and Samuel B. Stephens. 2025. "Breaking the Feedback Loop of β-Cell Failure: Insight into the Pancreatic β-Cell’s ER-Mitochondria Redox Balance" Cells 14, no. 6: 399. https://doi.org/10.3390/cells14060399
APA StyleZaher, A., & Stephens, S. B. (2025). Breaking the Feedback Loop of β-Cell Failure: Insight into the Pancreatic β-Cell’s ER-Mitochondria Redox Balance. Cells, 14(6), 399. https://doi.org/10.3390/cells14060399