
RAGE Cytosolic Partner Diaph1 Does Not Play an Essential Role in Diabetic Peripheral Neuropathy Progression

 , , ,
, , ,  , , and
, , and
Highlights
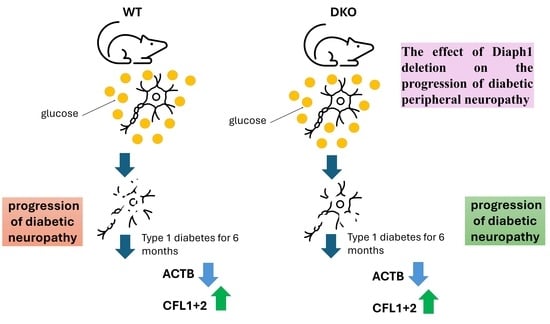
- Global deletion of Diaph1 disrupts beta-actin polymerization in the sciatic nerve during type 1 diabetes.
- We observed decreased nerve conduction velocity and abnormalities in sciatic nerve morphometry in diabetic Diaph1 knockout diabetic mice.
- Deletion of Diaph1 is insufficient to halt the progression of diabetic peripheral neuropathy in mice.
- However, we cannot unequivocally rule out that Diaph1 is an important switch role in the RAGE pathway and diabetic peripheral neuropathy.
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunofluorescence Staining
2.3. Morphometric Studies
2.4. Nerve Conduction Velocity (NCV)
2.5. Statistical Analysis
3. Results
3.1. Impact of Diaph1 Deletion on Weight and on Blood Glucose Levels
3.2. Amounts of ACTB, PFN1, CFL, and RhoA upon Deletion of Diaph1
3.3. Morphological Alterations in Sciatic Nerve
3.4. NCV Under Diaph1 Control
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RAGE | Receptor for advanced glycation end-products |
| AGE | Advanced glycation end-products |
| CFL | Cofilin |
| DPN | Diabetic peripheral neuropathy |
| Diaph1 | Diaphanous-related formin 1 |
| DKO | Diaph1 knockout mouse |
| FH1 | Formin homology 1 |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| mDia1 | Mammalian diaphanous |
| MNCV | Motor nerve conduction velocity |
| NCV | Nerve conduction velocity |
| PKC | Protein kinase C |
| ROI | Region of interest |
| SNCV | Sensory nerve conduction velocity |
| STZ | Streptozotocin |
| T1D | Type 1 diabetes |
| WT | Wild type |
References
- Juranek, J.; Mukherjee, K.; Kordas, B.; Załęcki, M.; Korytko, A.; Zglejc-Waszak, K.; Szuszkiewicz, J.; Banach, M. Role of RAGE in the Pathogenesis of Neurological Disorders. Neurosci. Bull. 2022, 38, 1248–1262. [Google Scholar] [CrossRef]
- Zglejc-Waszak, K.; Mukherjee, K.; Korytko, A.; Lewczuk, B.; Pomianowski, A.; Wojtkiewicz, J.; Banach, M.; Załęcki, M.; Nowicka, N.; Jarosławska, J.; et al. Novel insights into the nervous system affected by prolonged hyperglycemia. J. Mol. Med. 2023, 101, 1015–1028. [Google Scholar] [CrossRef]
- Zglejc-Waszak, K.; Mukherjee, K.; Juranek, J.K. The crosstalk between RAGE and DIAPH1 in neurological complications of diabetes: A review. Eur. J. Neurosci. 2024, 54, 5982–5999. [Google Scholar] [CrossRef]
- Theophall, G.G.; Premo, A.; Reverdatto, S.; Omojowolo, E.; Nazarian, P.; Burz, D.S.; Ramasamy, R.; Schmidt, A.M.; Shekhtman, A. Negative cooperativity regulates ligand activation of DIAPH1 and other diaphanous related formins. Commun. Biol. 2025, 8, 776. [Google Scholar] [CrossRef]
- Zglejc-Waszak, K.; Schmidt, A.M.; Juranek, J.K. The receptor for advanced glycation end products and its ligands’ expression in OVE26 diabetic sciatic nerve during the development of length-dependent neuropathy. Neuropathology 2023, 43, 84–94. [Google Scholar] [CrossRef]
- López-Díez, R.; Shen, X.; Daffu, G.; Khursheed, M.; Hu, J.; Song, F.; Rosario, R.; Xu, Y.; Li, Q.; Xi, X.; et al. Ager Deletion Enhances Ischemic Muscle Inflammation, Angiogenesis, and Blood Flow Recovery in Diabetic Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1536–1547. [Google Scholar] [CrossRef]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. RAGE/DIAPH1 and atherosclerosis through an evolving lens: Viewing the cell from the “Inside–Out”. Atherosclerosis 2024, 394, 117304. [Google Scholar] [CrossRef] [PubMed]
- Jaroslawska, J.; Korytko, A.; Zglejc-Waszak, K.; Antonowski, T.; Pomianowski, A.S.; Wasowicz, K.; Wojtkiewicz, J.; Juranek, J.K. Peripheral Neuropathy Presents Similar Symptoms and Pathological Changes in Both High-Fat Diet and Pharmacologically Induced Pre- and Diabetic Mouse Models. Life 2021, 11, 1267. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.K.; Geddis, M.S.; Song, F.; Zhang, J.; Garcia, J.; Rosario, R.; Yan, S.F.; Brannagan, T.H.; Schmidt, A.M. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes 2013, 62, 931–943. [Google Scholar] [CrossRef]
- Juranek, J.K.; Aleshin, A.; Rattigan, E.M.; Johnson, L.; Qu, W.; Song, F.; Ananthakrishnan, R.; Quadri, N.; Yan, S.D.; Ramasamy, R.; et al. Morphological Changes and Immunohistochemical Expression of RAGE and its Ligands in the Sciatic Nerve of Hyperglycemic Pig (Sus Scrofa). Biochem. Insights 2010, 2010, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Friedman, R.A.; Ramasamy, R.; D’Agati, V.; Schmidt, A.M. Deletion of the formin Diaph1 protects from structural and functional abnormalities in the murine diabetic kidney. Am. J. Physiol. Ren. Physiol. 2018, 315, F1601–F1612. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Yan, S.F.; Schmidt, A.M.; Shekhtman, A. Signal transduction in receptor for advanced glycation end products (RAGE): Solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J. Biol. Chem. 2012, 287, 5133–5144, https://doi.org/10.1074/jbc.M111.277731; Erratum in J. Biol. Chem. 2012, 287, 11283. [Google Scholar]
- Manigrasso, M.B.; Rabbani, P.; Egaña-Gorroño, L.; Quadri, N.; Frye, L.; Zhou, B.; Reverdatto, S.; Ramirez, L.S.; Dansereau, S.; Pan, J.; et al. Small-molecule antagonism of the interaction of the RAGE cytoplasmic domain with DIAPH1 reduces diabetic complications in mice. Sci. Transl. Med. 2021, 13, eabf7084. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. The RAGE/DIAPH1 Signaling Axis & Implications for the Pathogenesis of Diabetic Complications. Int. J. Mol. Sci. 2022, 23, 4579. [Google Scholar] [CrossRef]
- Yepuri, G.; Hasan, S.N.; Kumar, V.; Manigrasso, M.B.; Theophall, G.; Shekhtman, A.; Schmidt, A.M.; Ramasamy, R. Mechanistic underpinnings of AGEs-RAGE via DIAPH1 in ischemic, diabetic, and failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2025. epub ahead of print. [Google Scholar] [CrossRef]
- Schulz, A.; Walther, C.; Morrison, H.; Bauer, R. In vivo electrophysiological measurements on mouse sciatic nerves. J. Vis. Exp. 2014, 13, 51181. [Google Scholar] [CrossRef]
- Janusonis, S. Comparing two small samples with an unstable, treatment-independent baseline. J. Neurosci. Methods 2009, 179, 173–178. [Google Scholar] [CrossRef]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Madaule, P.; Reid, T.; Ishizaki, T.; Watanabe, G.; Kakizuka, A.; Saito, Y.; Nakao, K.; Jockusch, B.M.; Narumiya, S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997, 16, 3044–3056. [Google Scholar] [CrossRef]
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef]
- Li, D.; Sewer, M.B. RhoA and DIAPH1 mediate adrenocorticotropin-stimulated cortisol biosynthesis by regulating mitochondrial trafficking. Endocrinology 2010, 151, 4313–4323. [Google Scholar] [CrossRef]
- Otomo, T.; Otomo, C.; Tomchick, D.R.; Machius, M.; Rosen, M.K. Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol. Cell 2005, 18, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lu, L.; Chen, W.; Chen, H.; Xu, X.; Zheng, Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.S.; Medina, M.; Zuliani, C.; Di Nardo, A.; Witke, W.; Dotti, C.G. RhoA/ROCK regulation of neuritogenesis via profilin IIa-mediated control of actin stability. J. Cell Biol. 2003, 162, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Senatus, L.; Egaña-Gorroño, L.; López-Díez, R.; Bergaya, S.; Aranda, J.F.; Amengual, J.; Arivazhagan, L.; Manigrasso, M.B.; Yepuri, G.; Nimma, R.; et al. DIAPH1 mediates progression of atherosclerosis and regulates hepatic lipid metabolism in mice. Commun. Biol. 2023, 6, 280. [Google Scholar] [CrossRef]
- Elzinga, S.; Murdock, B.J.; Guo, K.; Hayes, J.M.; Tabbey, M.A.; Hur, J.; Feldman, E.L. Toll-like receptors and inflammation in metabolic neuropathy; a role in early versus late disease? Exp. Neurol. 2019, 320, 112967. [Google Scholar] [CrossRef]
- Inman, C.K.; Aljunaibi, A.; Koh, H.; Abdulle, A.; Ali, R.; Alnaeemi, A.; Al Zaabi, E.; Oumeziane, N.; Al Bastaki, M.; Al-Houqani, M.; et al. The AGE-RAGE axis in an Arab population: The United Arab Emirates Healthy Futures (UAEHFS) pilot study. J. Clin. Transl. Endocrinol. 2017, 10, 1–8. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights From Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Kim, W.; Hudson, B.I.; Moser, B.; Guo, J.; Rong, L.L.; Lu, Y.; Qu, W.; Lalla, E.; Lerner, S.; Chen, Y.; et al. Receptor for advanced glycation end products and its ligands: A journey from the complications of diabetes to its pathogenesis. Ann. N. Y. Acad. Sci. 2005, 1043, 553–561. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Bucciarelli, L.G.; Wendt, T.; Lee, L.K.; Hudson, B.I.; Stern, D.M.; Lalla, E.; Yan, S.D.; Rong, L.L.; et al. RAGE and its ligands: A lasting memory in diabetic complications? Diabetes Vasc. Dis. Res. 2004, 1, 10–20. [Google Scholar] [CrossRef]
- Azizoglu, Z.B.; Babayeva, R.; Haskologlu, Z.S.; Acar, M.B.; Ayaz-Guner, S.; Okus, F.Z.; Alsavaf, M.B.; Can, S.; Basaran, K.E.; Canatan, M.F.; et al. DIAPH1-Deficiency is Associated with Major T, NK and ILC Defects in Humans. J. Clin. Immunol. 2024, 44, 175, https://doi.org/10.1007/s10875-024-01777-8; Erratum in J. Clin. Immunol. 2024, 45, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fan, C.; Qie, T.; Fu, X.; Chen, X.; Wang, Y.; Wu, Y.; Fu, X.; Shi, K.; Yan, W.; et al. Diaph1 knockout inhibits mouse primordial germ cell proliferation and affects gonadal development. Reprod. Biol. Endocrinol. 2024, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Kaustio, M.; Nayebzadeh, N.; Hinttala, R.; Tapiainen, T.; Åström, P.; Mamia, K.; Pernaa, N.; Lehtonen, J.; Glumoff, V.; Rahikkala, E.; et al. Loss of DIAPH1 causes SCBMS, combined immunodeficiency, and mitochondrial dysfunction. J. Allergy Clin. Immunol. 2021, 148, 599–611, https://doi.org/10.1016/j.jaci.2020.12.656; Erratum in J. Allergy Clin. Immunol. 2021, 148, 1603. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, K.M.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Thiagarajan, D.; Sreejit, G.; Wang, L.; Zirpoli, H.; Aranda, J.F.; Alberts, A.S.; et al. The Formin, DIAPH1, is a Key Modulator of Myocardial Ischemia/Reperfusion Injury. eBioMedicine 2017, 26, 165–174. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | |||||
|---|---|---|---|---|---|
| Antigen | Code | Species | Working Dilution | Supplier | |
| Immunofluorescence staining | |||||
| Sciatic nerve | |||||
| ACTB | 251006 | Chicken | 1:200 | Synaptic System, Germany | |
| PFN1 | Ab232020 | Rabbit | 1:100 | Abcam, CA, UK | |
| CFL1 + 2 | Ab131519 | 1:100 | |||
| RhoA | Ab187027 | 1:100 | |||
| Secondary antibody | |||||
| Immunofluorescence staining | |||||
| Reagents | Code | Working dilution | Supplier | ||
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 594 | A32740 | 1:2000 | ThermoFisher, Oxford, UK | ||
| Goat anti-Chicken IgY, Alexa Fluor 488 | A-11039 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zglejc-Waszak, K.; Kordas, B.; Korytko, A.; Pomianowski, A.; Lewczuk, B.; Wojtkiewicz, J.; Wąsowicz, K.; Babińska, I.; Mukherjee, K.; Juranek, J. RAGE Cytosolic Partner Diaph1 Does Not Play an Essential Role in Diabetic Peripheral Neuropathy Progression. Cells 2025, 14, 1635. https://doi.org/10.3390/cells14201635
Zglejc-Waszak K, Kordas B, Korytko A, Pomianowski A, Lewczuk B, Wojtkiewicz J, Wąsowicz K, Babińska I, Mukherjee K, Juranek J. RAGE Cytosolic Partner Diaph1 Does Not Play an Essential Role in Diabetic Peripheral Neuropathy Progression. Cells. 2025; 14(20):1635. https://doi.org/10.3390/cells14201635
Chicago/Turabian StyleZglejc-Waszak, Kamila, Bernard Kordas, Agnieszka Korytko, Andrzej Pomianowski, Bogdan Lewczuk, Joanna Wojtkiewicz, Krzysztof Wąsowicz, Izabella Babińska, Konark Mukherjee, and Judyta Juranek. 2025. "RAGE Cytosolic Partner Diaph1 Does Not Play an Essential Role in Diabetic Peripheral Neuropathy Progression" Cells 14, no. 20: 1635. https://doi.org/10.3390/cells14201635
APA StyleZglejc-Waszak, K., Kordas, B., Korytko, A., Pomianowski, A., Lewczuk, B., Wojtkiewicz, J., Wąsowicz, K., Babińska, I., Mukherjee, K., & Juranek, J. (2025). RAGE Cytosolic Partner Diaph1 Does Not Play an Essential Role in Diabetic Peripheral Neuropathy Progression. Cells, 14(20), 1635. https://doi.org/10.3390/cells14201635