



Early Pharmacodynamic Changes Measured Using RNA Sequencing of Peripheral Blood from Patients in a Phase I Study with Mitazalimab, a Potent CD40 Agonistic Monoclonal Antibody

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Design
2.2. RNA Sequencing of Blood Samples
2.3. Cytokine Analysis
2.4. Statistical Testing
3. Results
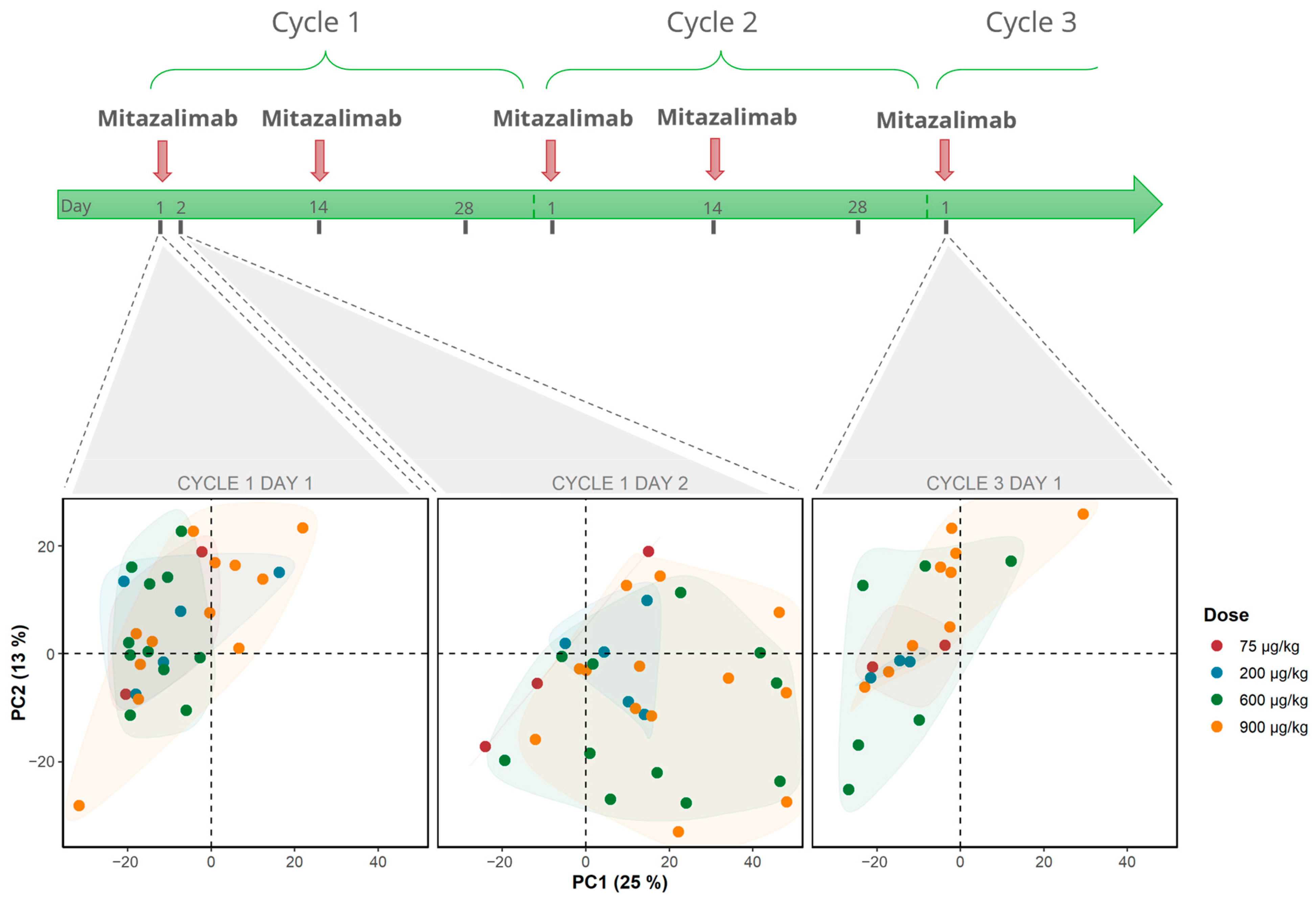
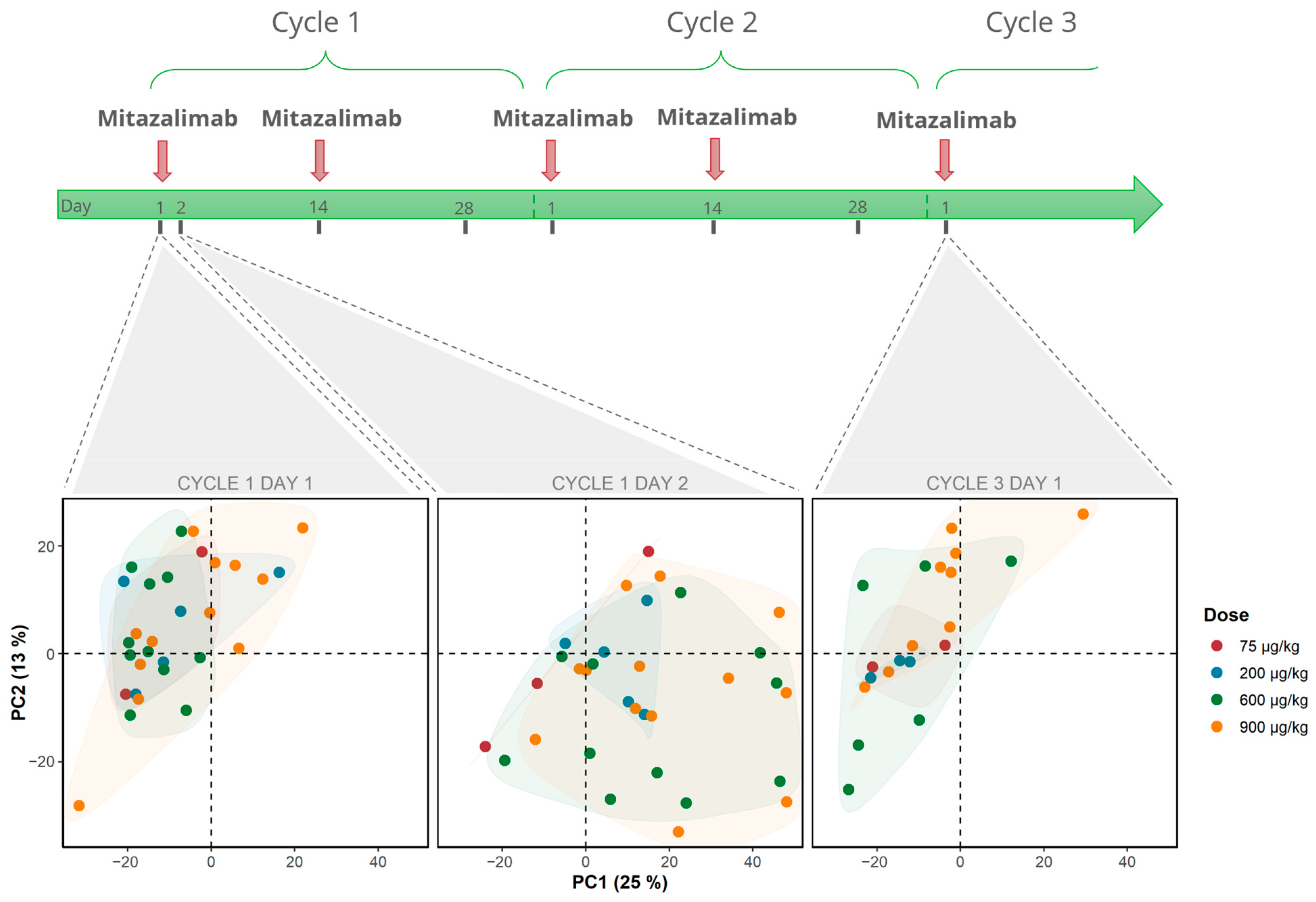
3.1. Mitazalimab Induces Transient Peripheral Transcriptomic Alterations in Patients with Advanced Solid Tumors
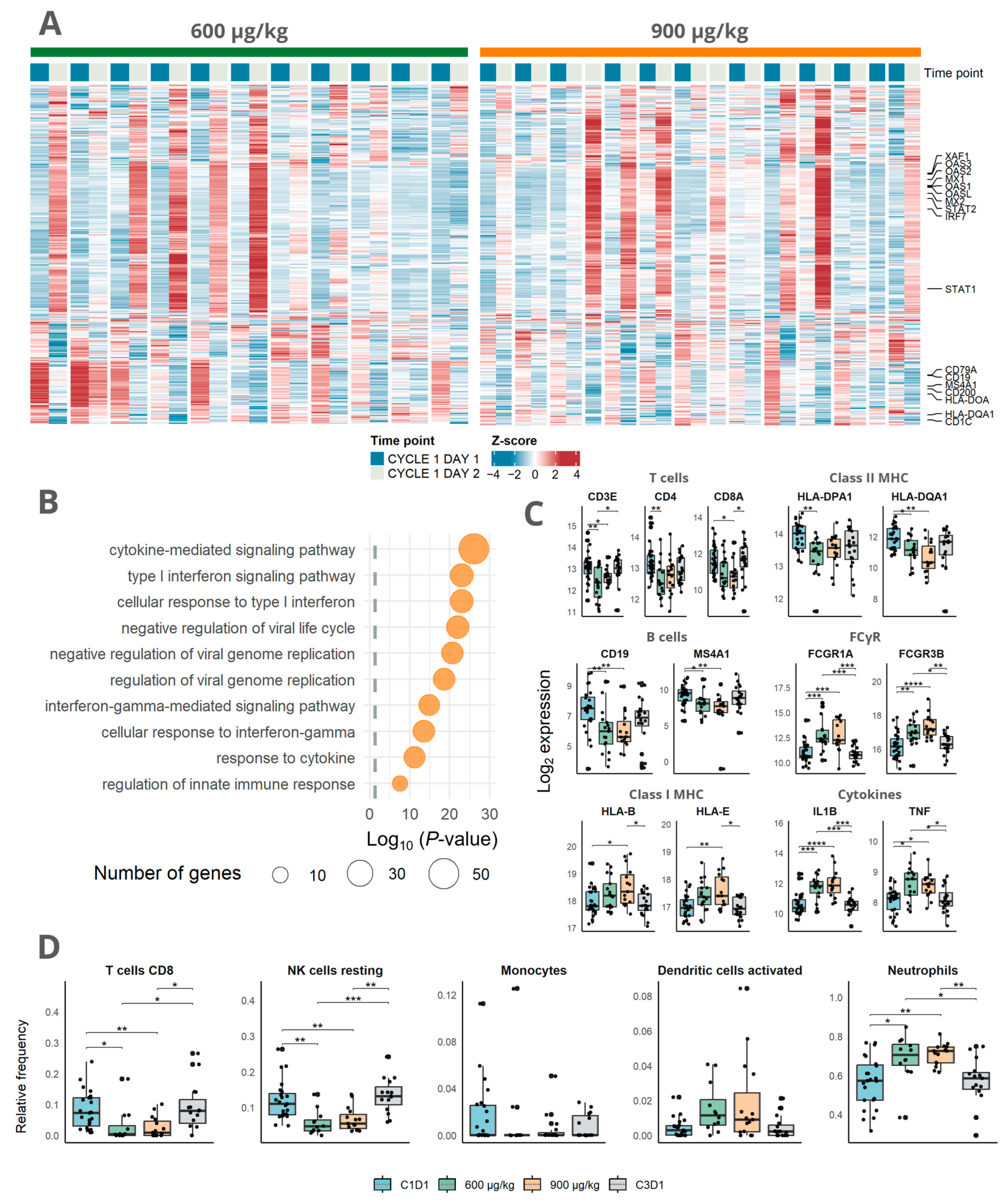
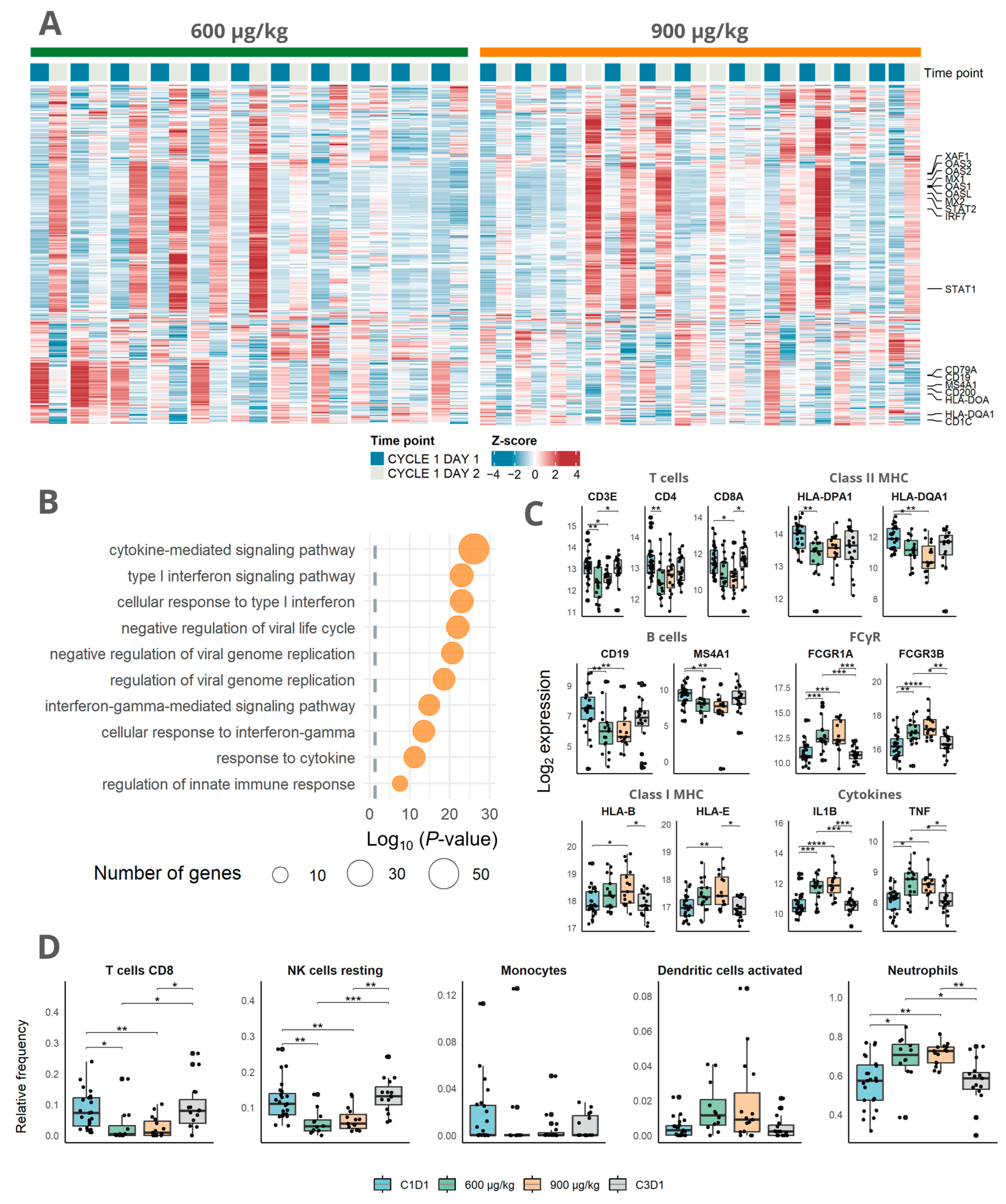
3.2. Immune Activation Induced by Mitazalimab Is Detectable in Blood Transcriptome
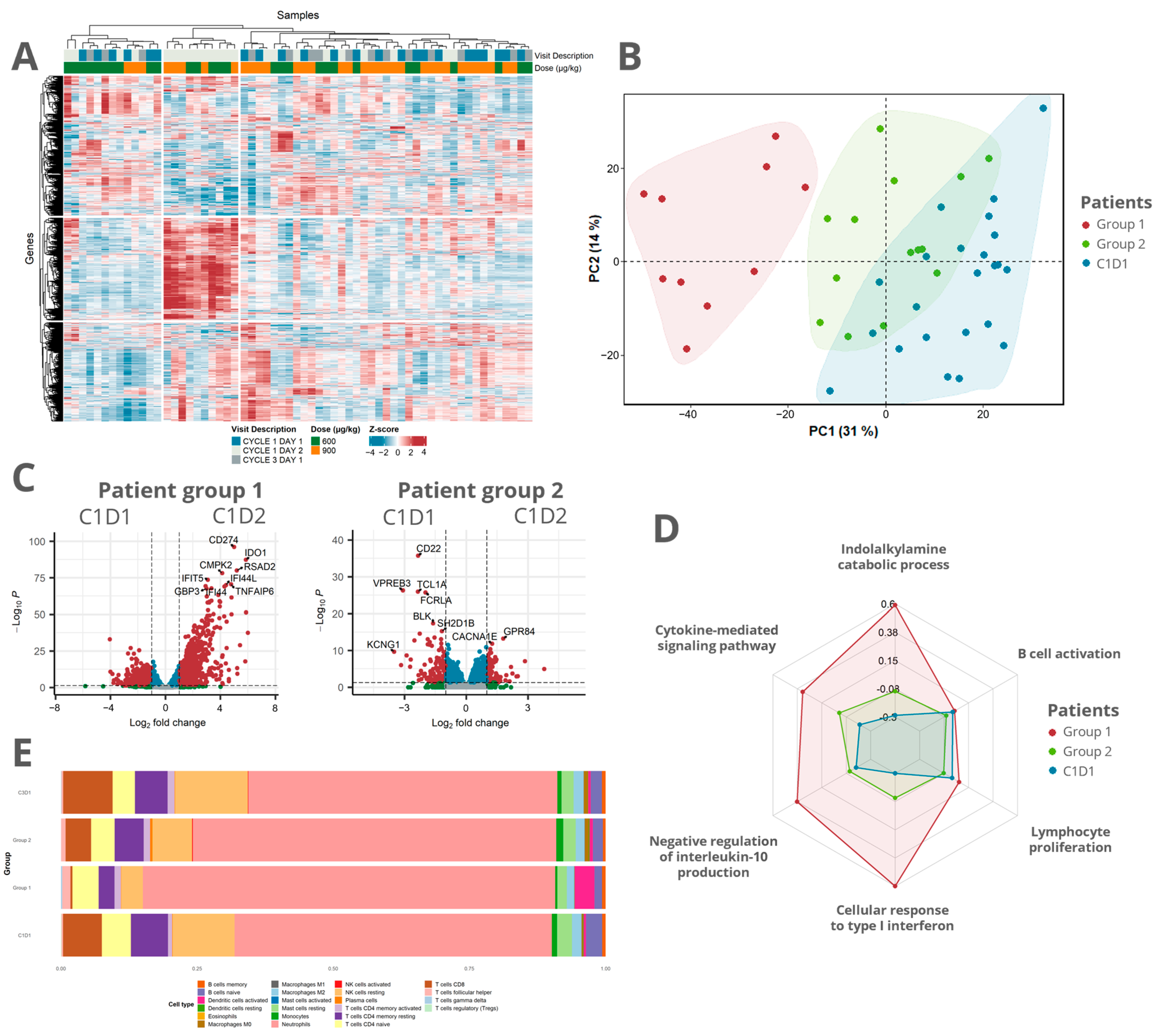
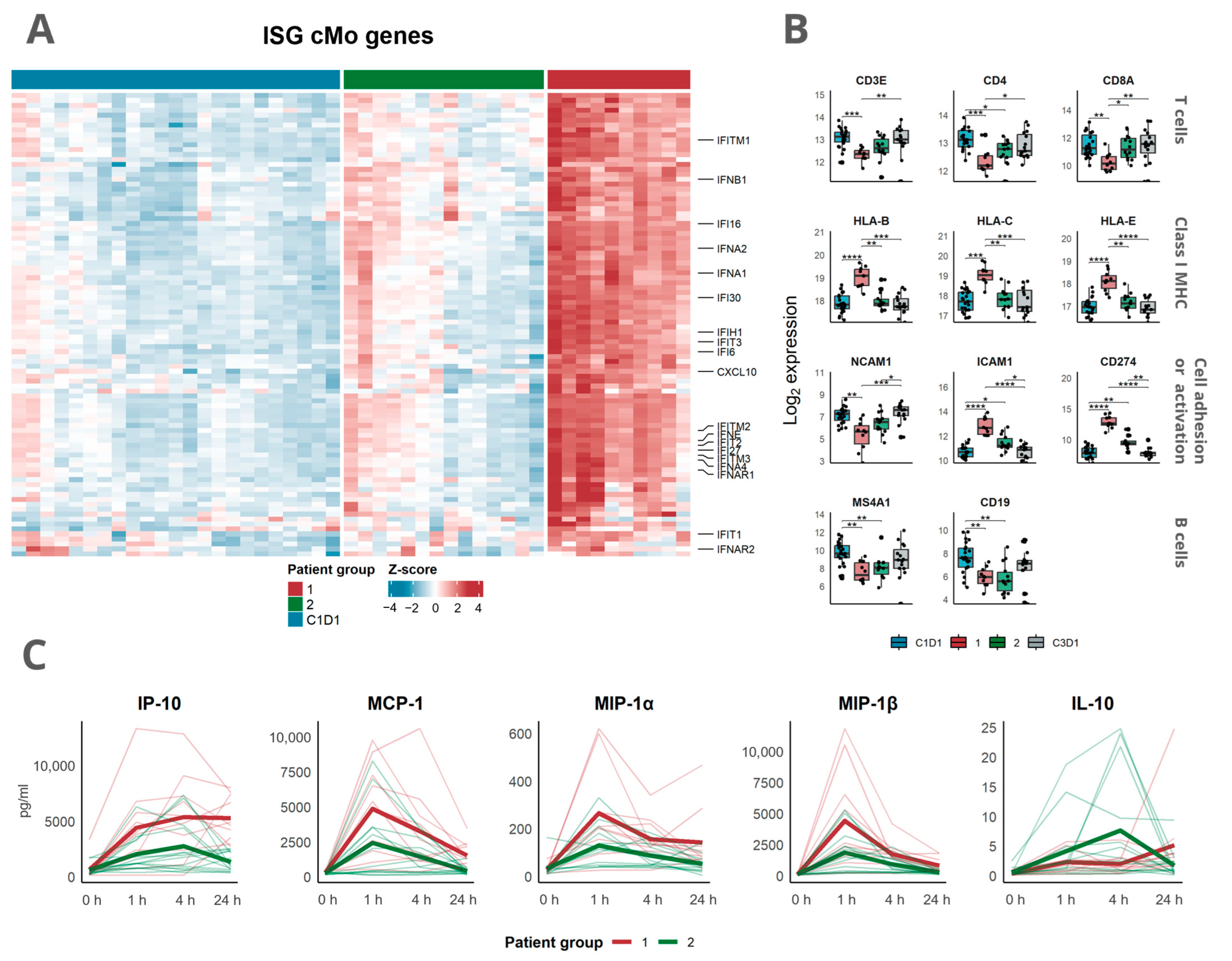
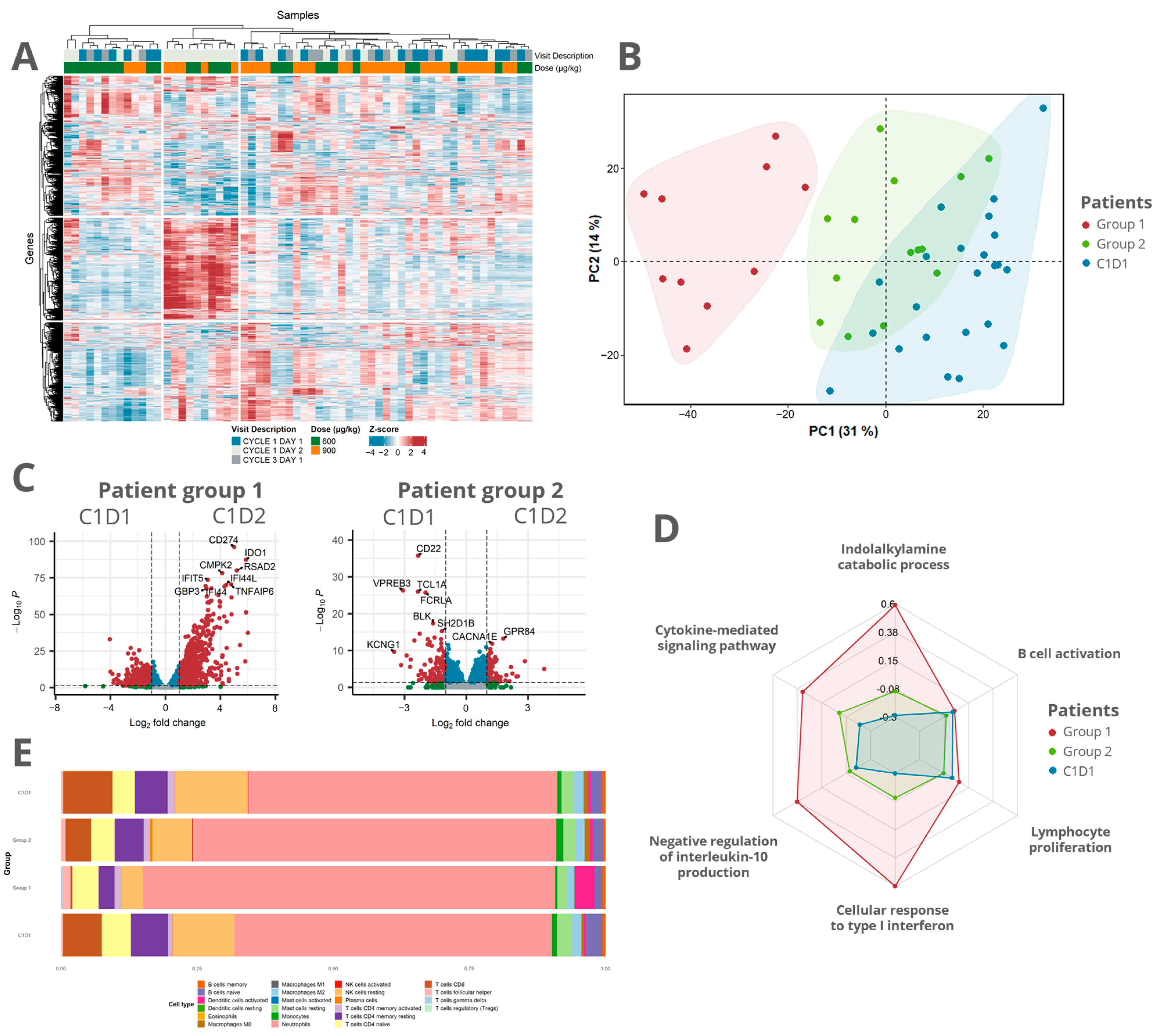
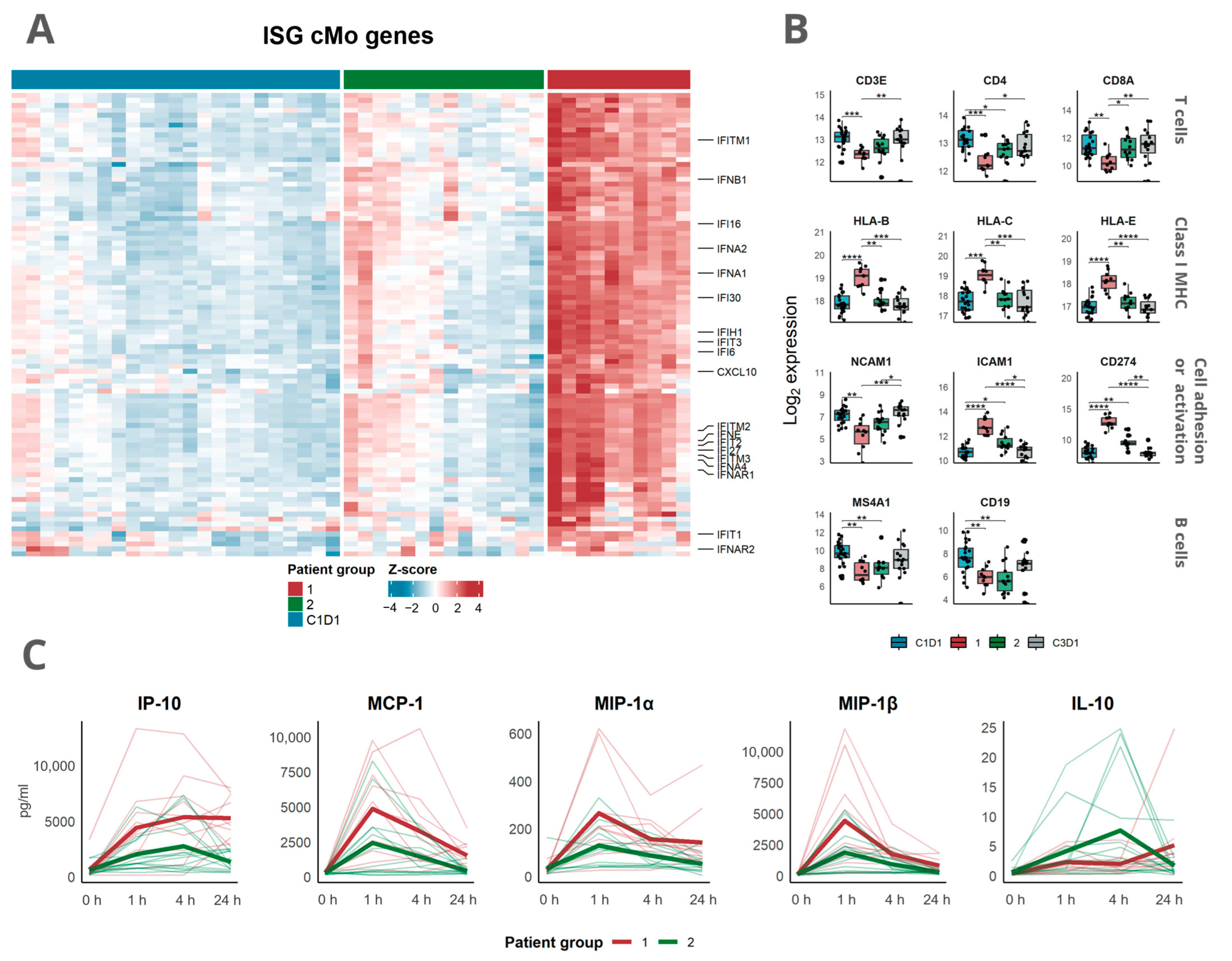
3.3. Transcriptomic Analysis Revealed a Distinct Response Pattern in a Subset of the Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The Association between CD8+ Tumor-Infiltrating Lymphocytes and the Clinical Outcome of Cancer Immunotherapy: A Systematic Review and Meta-Analysis. eClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Bazan, F.; Blanchard, D.; Briè, F.; Galizzi, J.P. The CD40 Antigen and Its Ligand. Annu. Rev. Immunol. 1994, 12, 881–922. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.Y.; Clark, E.A. The Role of CD40 and CD40L in Dendritic Cells. Semin. Immunol. 2009, 21, 265–272. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef]
- Enell Smith, K.; Deronic, A.; Hägerbrand, K.; Norlén, P.; Ellmark, P. Rationale and Clinical Development of CD40 Agonistic Antibodies for Cancer Immunotherapy. Expert Opin. Biol. Ther. 2021, 21, 1635–1646. [Google Scholar] [CrossRef]
- Yu, R.; Zhu, B.; Chen, D. Type I Interferon-Mediated Tumor Immunity and Its Role in Immunotherapy. Cell. Mol. Life Sci. 2022, 79, 191. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning Cold Tumors Hot: From Molecular Mechanisms to Clinical Applications. Trends Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFN-γ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef]
- Djureinovic, D.; Wang, M.; Kluger, H.M. Agonistic CD40 Antibodies in Cancer Treatment. Cancers 2021, 13, 1302. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Sznol, M.; Shaheen, M.; Berciano-Guerrero, M.-Á.; Felip, E.; Rodríguez-Abreu, D.; Arance, A.M.; Boni, V.; Linette, G.; Schuchter, L.; et al. 389 Phase II of CD40 Agonistic Antibody Sotigalimab (APX005M) in Combination with Nivolumab in Subjects with Metastatic Melanoma with Confirmed Disease Progression on Anti-PD-1 Therapy. J. Immunother. Cancer 2021, 9, A422. [Google Scholar] [CrossRef]
- Mangsbo, S.M.; Broos, S.; Fletcher, E.; Veitonmäki, N.; Furebring, C.; Dahlén, E.; Norlén, P.; Lindstedt, M.; Tötterman, T.H.; Ellmark, P. The Human Agonistic CD40 Antibody ADC-1013 Eradicates Bladder Tumors and Generates T-Cell–Dependent Tumor Immunity. Clin. Cancer Res. 2015, 21, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Irenaeus, S.M.M.; Nielsen, D.; Ellmark, P.; Yachnin, J.; Deronic, A.; Nilsson, A.; Norlén, P.; Veitonmäki, N.; Wennersten, C.S.; Ullenhag, G.J. First-in-Human Study with Intratumoral Administration of a CD40 Agonistic Antibody, ADC-1013, in Advanced Solid Malignancies. Int. J. Cancer 2019, 145, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Perets, R.; Peretz-Yablonski, T.; Fourneau, N.; Girgis, S.; Guo, Y.; Hellemans, P.; Verona, R.; Pendás, N.; Xia, Q.; et al. A Phase 1 Study of Intravenous Mitazalimab, a CD40 Agonistic Monoclonal Antibody, in Patients with Advanced Solid Tumors. Investig. New Drugs 2022, 41, 93–104. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical Activity and Immune Modulation in Cancer Patients Treated with CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, with or without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Deronic, A.; Nilsson, A.; Thagesson, M.; Werchau, D.; Smith, K.E.; Ellmark, P. The Human Anti-CD40 Agonist Antibody Mitazalimab (ADC-1013; JNJ-64457107) Activates Antigen-Presenting Cells, Improves Expansion of Antigen-Specific T Cells, and Enhances Anti-Tumor Efficacy of a Model Cancer Vaccine In Vivo. Cancer Immunol. Immunother. 2021, 70, 3629–3642. [Google Scholar] [CrossRef]
- Pantano, L.; Hutchinson, J.; Barrera, V.; Piper, M.; Khetani, R.; Daily, K.; Malai Perumal, T.; Kirchner, R.; Steinbaugh, M. DEGreport: Report of DEG Analysis. Available online: http://lpantano.github.io/DEGreport/ (accessed on 18 September 2023).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Blighe, K. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling 2023. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 18 September 2023).
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining Cell Type Abundance and Expression from Bulk Tissues with Digital Cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R Package for Assessing the Uncertainty in Hierarchical Clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Mulder, K.; Patel, A.A.; Kong, W.T.; Piot, C.; Halitzki, E.; Dunsmore, G.; Khalilnezhad, S.; Irac, S.E.; Dubuisson, A.; Chevrier, M.; et al. Cross-Tissue Single-Cell Landscape of Human Monocytes and Macrophages in Health and Disease. Immunity 2021, 54, 1883–1900.e5. [Google Scholar] [CrossRef]
- Rüter, J.; Antonia, S.J.; Burris, H.A.; Huhn, R.D.; Vonderheide, R.H. Immune Modulation with Weekly Dosing of an Agonist CD40 Antibody in a Phase I Study of Patients with Advanced Solid Tumors. Cancer Biol. Ther. 2010, 10, 983–993. [Google Scholar] [CrossRef]
- Bensinger, W.; Maziarz, R.T.; Jagannath, S.; Spencer, A.; Durrant, S.; Becker, P.S.; Ewald, B.; Bilic, S.; Rediske, J.; Baeck, J.; et al. A Phase 1 Study of Lucatumumab, a Fully Human Anti-CD40 Antagonist Monoclonal Antibody Administered Intravenously to Patients with Relapsed or Refractory Multiple Myeloma. Br. J. Haematol. 2012, 159, 58–66. [Google Scholar] [CrossRef]
- Johnson, P.; Challis, R.; Chowdhury, F.; Gao, Y.; Harvey, M.; Geldart, T.; Kerr, P.; Chan, C.; Smith, A.; Steven, N.; et al. Clinical and Biological Effects of an Agonist Anti-CD40 Antibody: A Cancer Research UK Phase I Study. Clin. Cancer Res. 2015, 21, 1321–1328. [Google Scholar] [CrossRef]
- Yang, X.; Kui, L.; Tang, M.; Li, D.; Wei, K.; Chen, W.; Miao, J.; Dong, Y. High-Throughput Transcriptome Profiling in Drug and Biomarker Discovery. Front. Genet. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. RNA Sequencing: Advances, Challenges and Opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Strohm, L.; Ubbens, H.; Münzel, T.; Daiber, A.; Daub, S. Role of CD40(L)-TRAF Signaling in Inflammation and Resolution—A Double-Edged Sword. Front. Pharmacol. 2022, 13, 995061. [Google Scholar] [CrossRef] [PubMed]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E.-H. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.G.; Arber, D.A. CD79: A Review. Appl. Immunohistochem. Mol. Morphol. AIMM 2001, 9, 97–106. [Google Scholar] [CrossRef]
- Pende, D.; Falco, M.; Vitale, M.; Cantoni, C.; Vitale, C.; Munari, E.; Bertaina, A.; Moretta, F.; Del Zotto, G.; Pietra, G.; et al. Killer Ig-Like Receptors (KIRs): Their Role in NK Cell Modulation and Developments Leading to Their Clinical Exploitation. Front. Immunol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Sheikh, N.A.; Jones, L.A. CD54 Is a Surrogate Marker of Antigen Presenting Cell Activation. Cancer Immunol. Immunother. 2008, 57, 1381–1390. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- de Saint-Vis, B.; Vincent, J.; Vandenabeele, S.; Vanbervliet, B.; Pin, J.-J.; Aït-Yahia, S.; Patel, S.; Mattei, M.-G.; Banchereau, J.; Zurawski, S.; et al. A Novel Lysosome-Associated Membrane Glycoprotein, DC-LAMP, Induced upon DC Maturation, Is Transiently Expressed in MHC Class II Compartment. Immunity 1998, 9, 325–336. [Google Scholar] [CrossRef]
- Bai, M.; Grieshaber-Bouyer, R.; Wang, J.; Schmider, A.B.; Wilson, Z.S.; Zeng, L.; Halyabar, O.; Godin, M.D.; Nguyen, H.N.; Levescot, A.; et al. CD177 Modulates Human Neutrophil Migration through Activation-Mediated Integrin and Chemoreceptor Regulation. Blood 2017, 130, 2092–2100. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Number of Patients | |
|---|---|---|
| n | 38 * | |
| Age, years (median (range)) | 56 (18–77) | |
| Sex, n (%) | Female | 19 (50.0) |
| Male | 19 (50.0) | |
| Corticosteroid pretreatment, n (%) | No | 33 (86.8) |
| Yes | 5 (13.2) | |
| Dose (µg/kg), n (%) | 75 | 3 (7.9) |
| 200 | 5 (13.2) | |
| 600 | 16 (42.1) | |
| 900 | 14 (36.8) | |
| Cancer type, n (%) | Adrenal | 1 (2.6) |
| Breast | 4 (10.5) | |
| Cervical | 2 (5.3) | |
| Cholangiocarcinoma | 3 (7.9) | |
| Choroidal melanoma | 1 (2.6) | |
| Colorectal | 3 (7.9) | |
| Melanoma | 2 (5.3) | |
| Neuroendocrine | 1 (2.6) | |
| NSCLC | 3 (7.9) | |
| Pancreas | 3 (7.9) | |
| Rectal cancer | 1 (2.6) | |
| Renal cancer | 1 (2.6) | |
| Salivary gland | 3 (7.9) | |
| Sarcoma | 5 (13.2) | |
| Testicular | 1 (2.6) | |
| Thymus | 3 (7.9) | |
| Thyroid | 1 (2.6) | |
| Prior lines of treatment, n (%) | 0–4 | 30 (78.9) |
| >4 | 8 (21.1) | |
| Median (range) | 3 (0–9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, H.; Sobti, A.; Jimenez, D.G.; de Coaña, Y.P.; Ambarkhane, S.V.; Hägerbrand, K.; Smith, K.E.; Lindstedt, M.; Ellmark, P. Early Pharmacodynamic Changes Measured Using RNA Sequencing of Peripheral Blood from Patients in a Phase I Study with Mitazalimab, a Potent CD40 Agonistic Monoclonal Antibody. Cells 2023, 12, 2365. https://doi.org/10.3390/cells12192365
Andersson H, Sobti A, Jimenez DG, de Coaña YP, Ambarkhane SV, Hägerbrand K, Smith KE, Lindstedt M, Ellmark P. Early Pharmacodynamic Changes Measured Using RNA Sequencing of Peripheral Blood from Patients in a Phase I Study with Mitazalimab, a Potent CD40 Agonistic Monoclonal Antibody. Cells. 2023; 12(19):2365. https://doi.org/10.3390/cells12192365
Chicago/Turabian StyleAndersson, Hampus, Aastha Sobti, David Gomez Jimenez, Yago Pico de Coaña, Sumeet Vijay Ambarkhane, Karin Hägerbrand, Karin Enell Smith, Malin Lindstedt, and Peter Ellmark. 2023. "Early Pharmacodynamic Changes Measured Using RNA Sequencing of Peripheral Blood from Patients in a Phase I Study with Mitazalimab, a Potent CD40 Agonistic Monoclonal Antibody" Cells 12, no. 19: 2365. https://doi.org/10.3390/cells12192365
APA StyleAndersson, H., Sobti, A., Jimenez, D. G., de Coaña, Y. P., Ambarkhane, S. V., Hägerbrand, K., Smith, K. E., Lindstedt, M., & Ellmark, P. (2023). Early Pharmacodynamic Changes Measured Using RNA Sequencing of Peripheral Blood from Patients in a Phase I Study with Mitazalimab, a Potent CD40 Agonistic Monoclonal Antibody. Cells, 12(19), 2365. https://doi.org/10.3390/cells12192365