
Inflammatory Caspase Activity Mediates HMGB1 Release and Differentiation in Myoblasts Affected by Peripheral Arterial Disease

 , , , and
, , , and 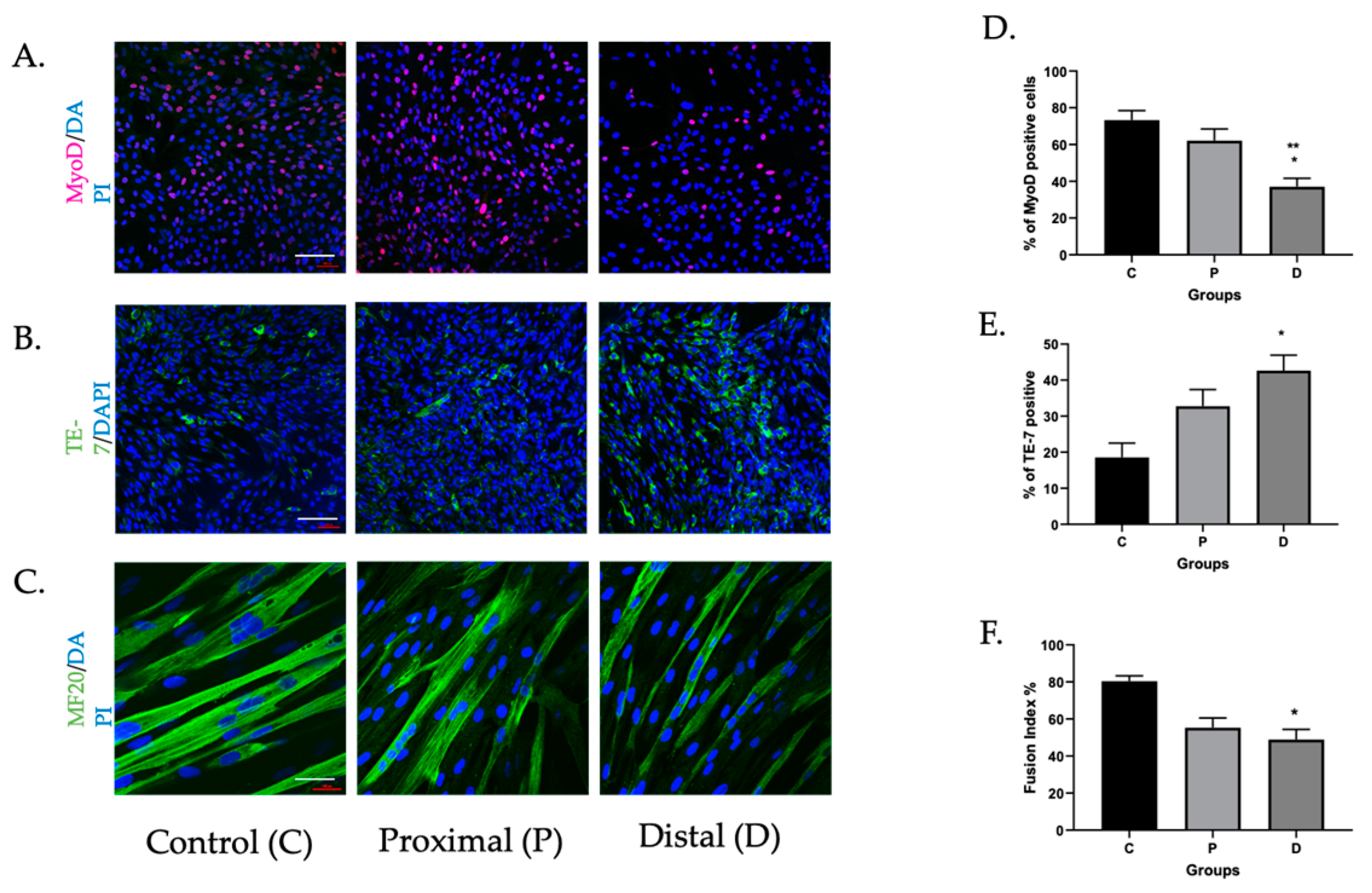{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects for Myoblast Isolation
2.2. Human Subjects for Core Needle Biopsies
2.3. Myoblast Harvest
2.4. Core-Needle Biopsies
2.5. Myogenicity Studies
2.6. Real-Time Polymerase Chain Reaction (RT-PCR)
2.7. Immunohistochemistry and Cytochemistry
2.8. Caspase-1 Activity Fluorometric Assay
2.9. Western Blot
2.10. Statistical Analysis
3. Results
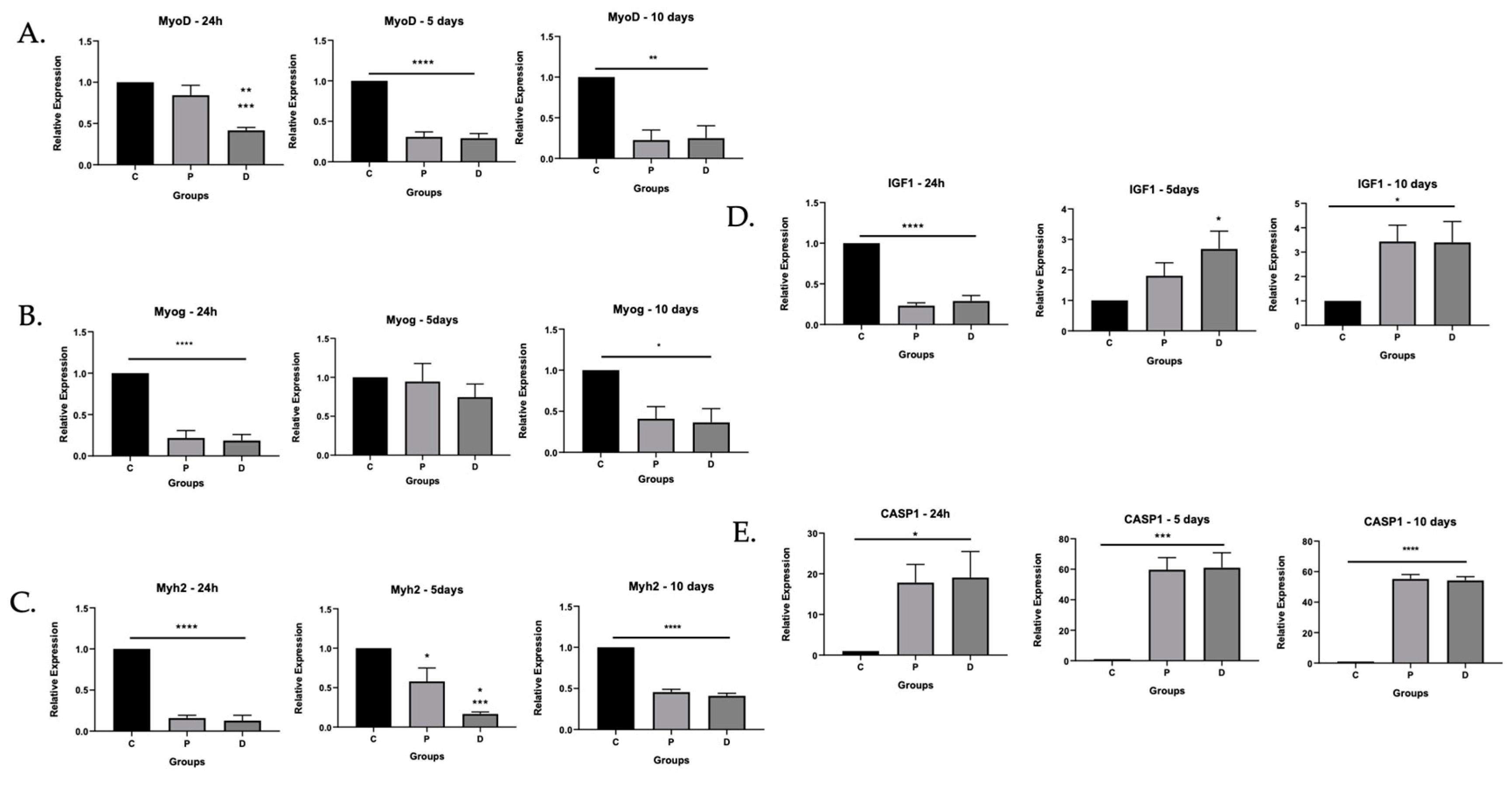
3.1. Myoblasts in PAD Were Viable but Their Differentiation Capacity Was Reduced
3.1.1. Myoblasts Can Be Harvested from Ischemic Muscle Tissue
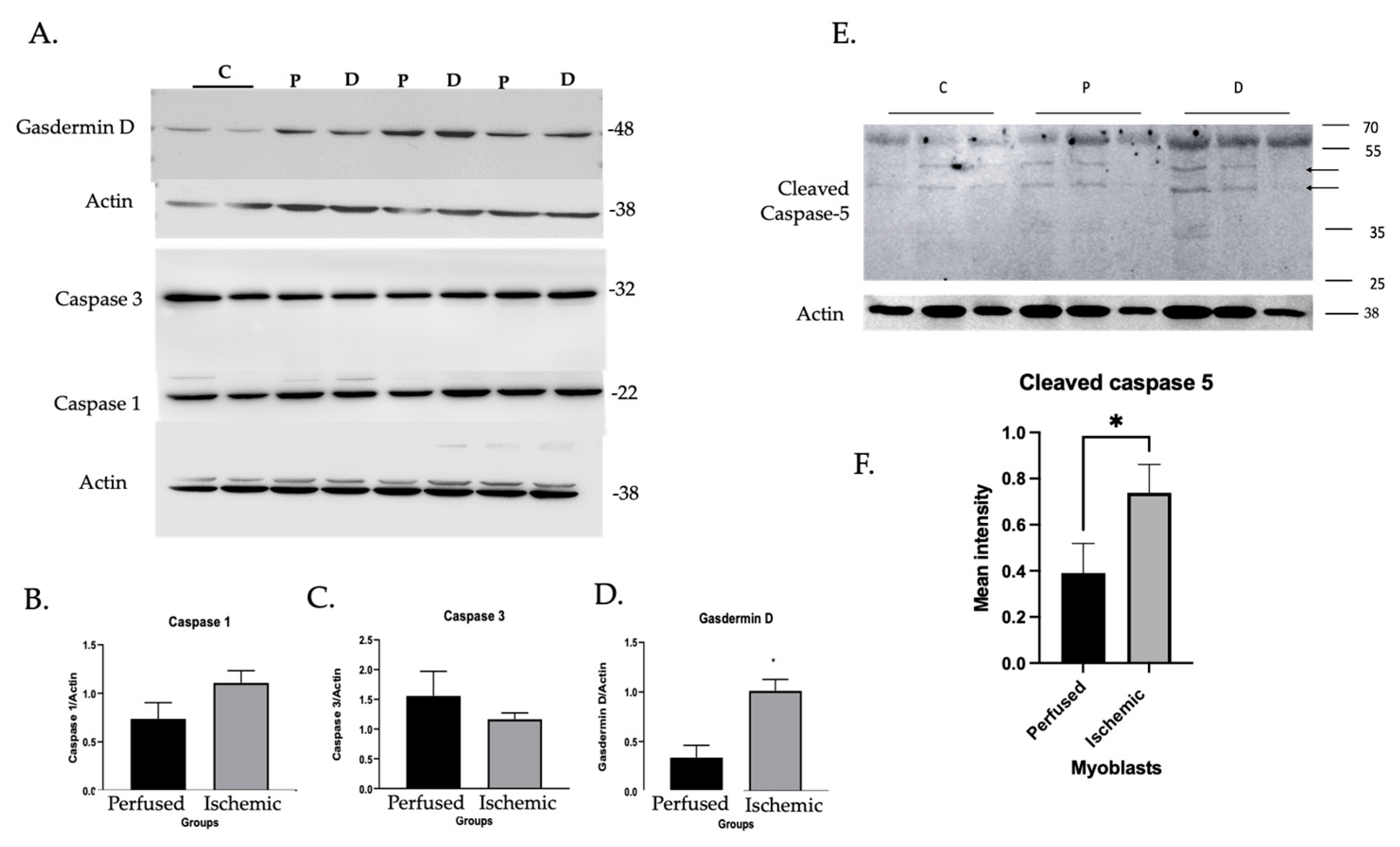
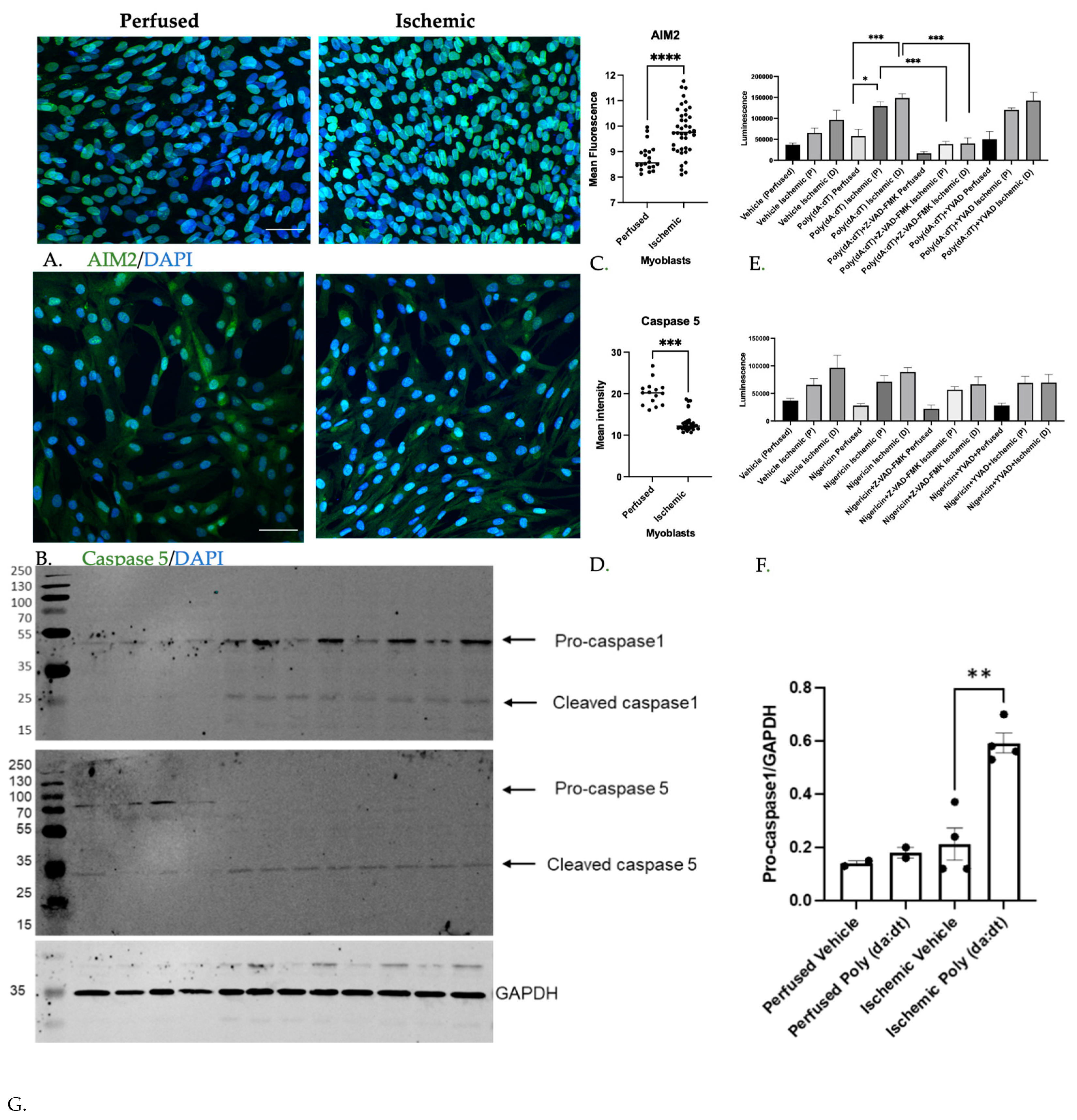
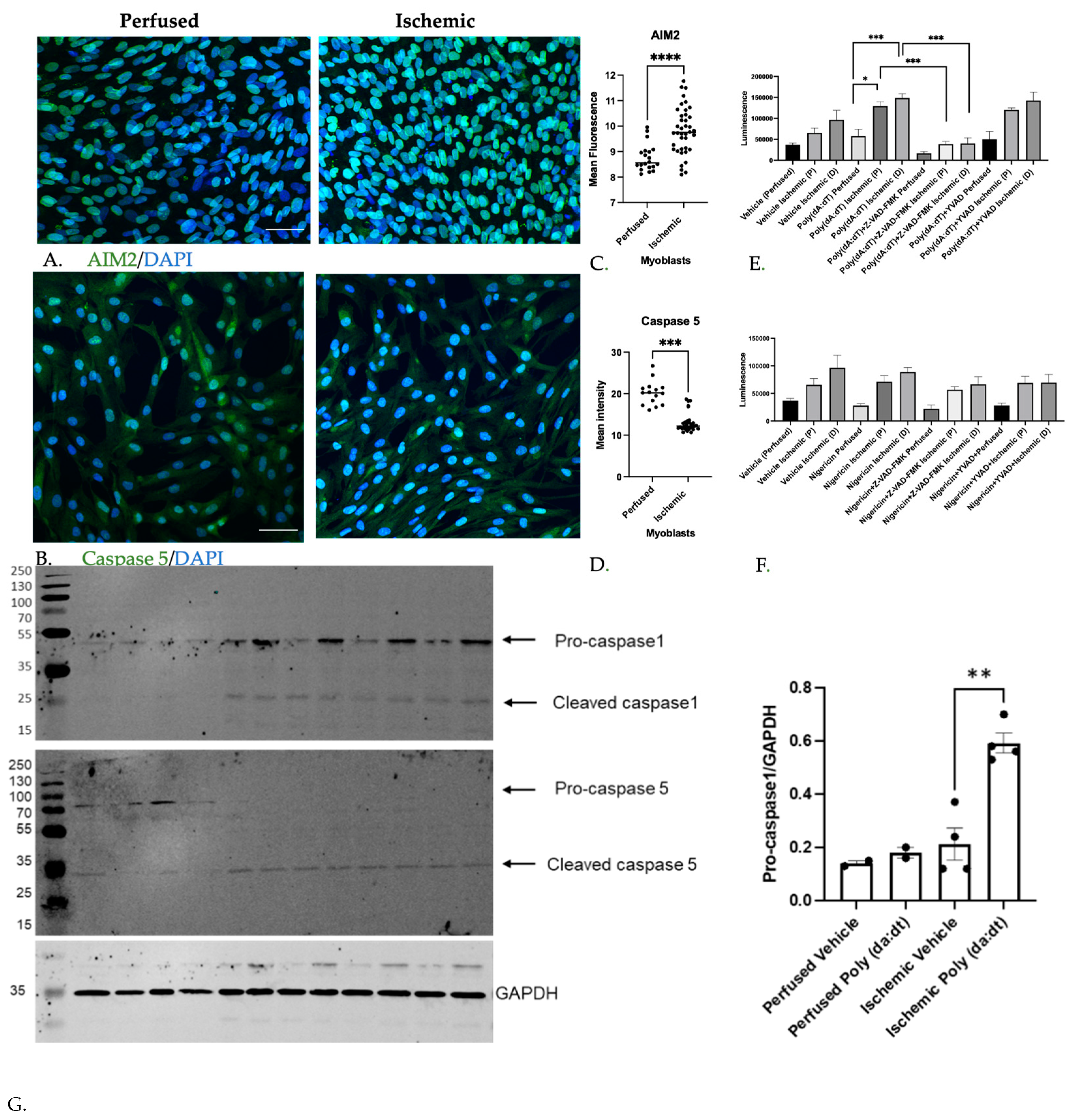
3.1.2. Inflammatory Caspase Activity in Ischemic Myoblasts Responded to AIM2 Agonists
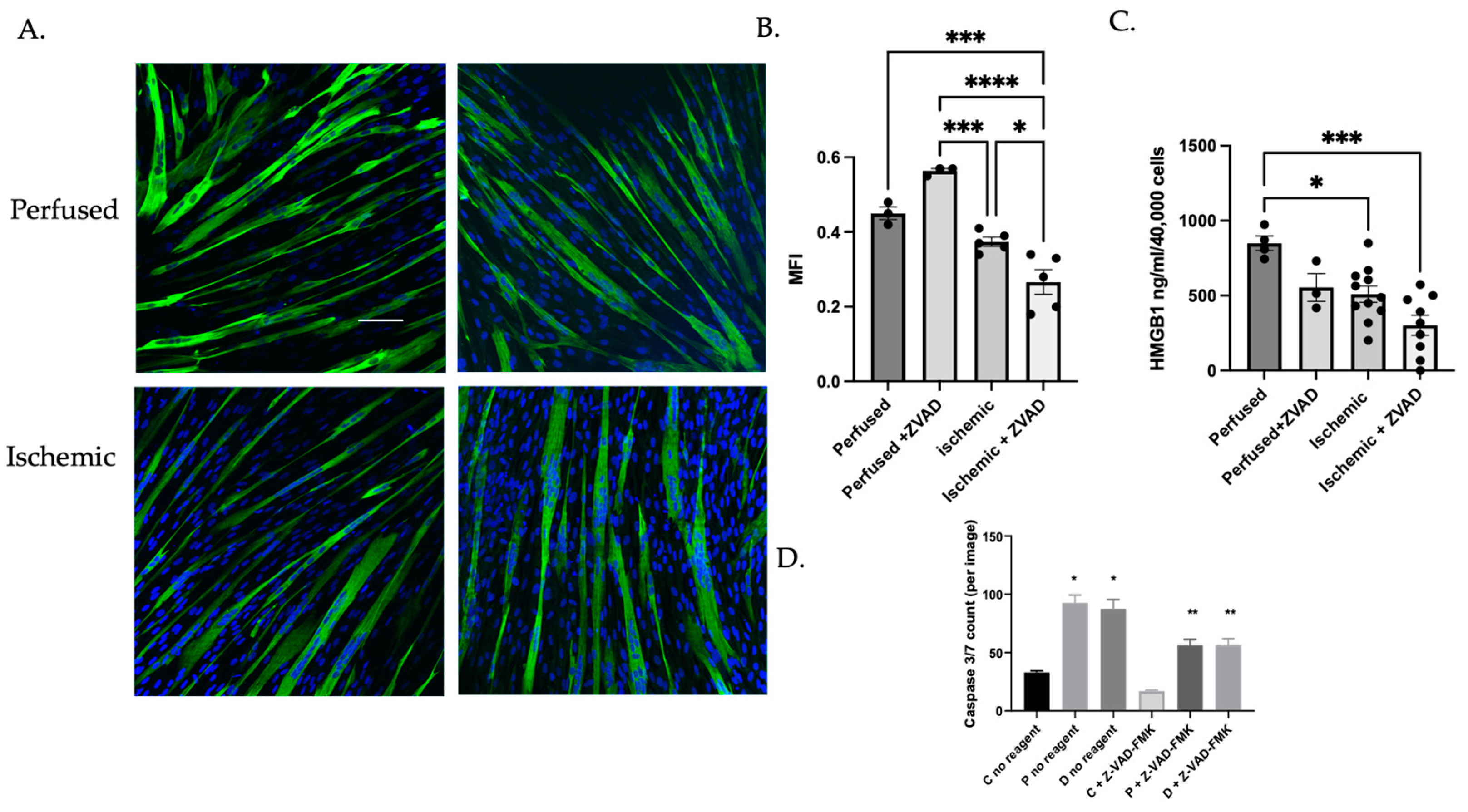
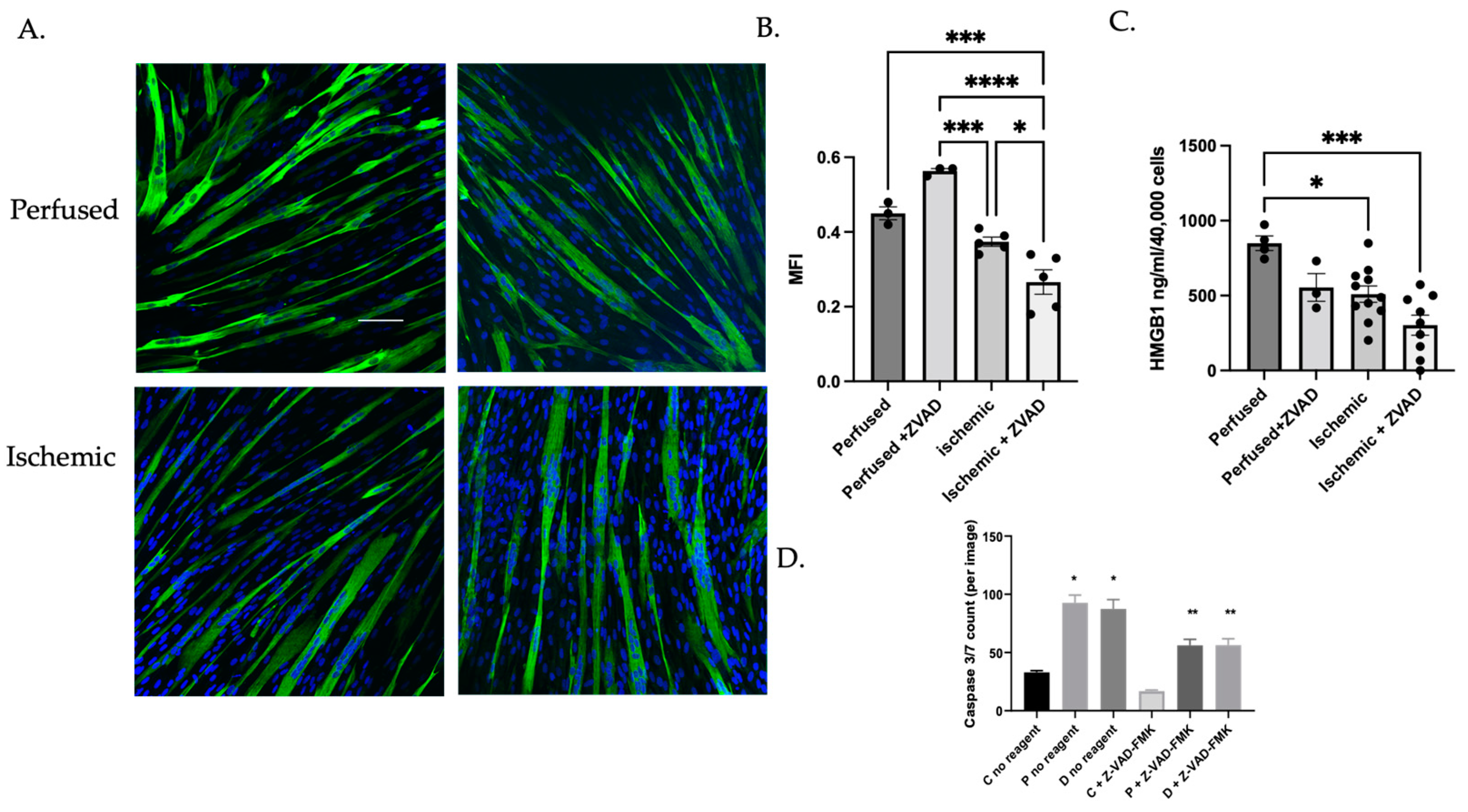
3.1.3. Caspase Inhibition Reduced Myocyte Differentiation in Ischemic Myoblasts
3.1.4. MuSC Released HMGB1 in a Caspase-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selvin, E.; Erlinger, T.P. Prevalence of and Risk Factors for Peripheral Arterial Disease in the United States: Results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 2004, 110, 738–743. [Google Scholar] [CrossRef] [Green Version]
- Koutakis, P.; Miserlis, D.; Myers, S.A.; Kim, J.K.; Zhu, Z.; Papoutsi, E.; Swanson, S.A.; Haynatzki, G.; Ha, D.M.; Carpenter, L.A.; et al. Abnormal Accumulation of Desmin in Gastrocnemius Myofibers of Patients with Peripheral Artery Disease: Associations with Altered Myofiber Morphology and Density, Mitochondrial Dysfunction and Impaired Limb Function. J. Histochem. Cytochem. 2015, 63, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutakis, P.; Myers, S.A.; Cluff, K.; Ha, D.M.; Haynatzki, G.; McComb, R.D.; Uchida, K.; Miserlis, D.; Papoutsi, E.; Johanning, J.M.; et al. Abnormal Myofiber Morphology and Limb Dysfunction in Claudication. J. Surg. Res. 2015, 196, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.N.; Hasenkamp, R.M.; Pipinos, I.I.; Johanning, J.M.; Stergiou, N.; DeSpiegelaere, H.K.; Chien, J.H.; Myers, S.A. Muscle Strength and Control Characteristics Are Altered by Peripheral Artery Disease. J. Vasc. Surg. 2017, 66, 178–186.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addison, O.; Prior, S.J.; Kundi, R.; Serra, M.C.; Katzel, L.I.; Gardner, A.W.; Ryan, A.S. Sarcopenia in Peripheral Arterial Disease: Prevalence and Effect on Functional Status. Arch. Phys. Med. Rehabil. 2018, 99, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Parmenter, B.J.; Raymond, J.; Dinnen, P.J.; Lusby, R.J.; Singh, M.A.F. Preliminary Evidence That Low Ankle-Brachial Index Is Associated with Reduced Bilateral Hip Extensor Strength and Functional Mobility in Peripheral Arterial Disease. J. Vasc. Surg. 2013, 57, 963–973.e1. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The Nlrp3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viganò, E.; Diamond, C.; Spreafico, R.; Balachander, A.; Sobota, R.; Mortellaro, A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, U.; Ferrari, R.; Cui, X.; Pius, A.; Sahu, A.; Reynolds, M.; Liao, H.; Sun, P.; Shinde, S.; Ambrosio, F.; et al. Caspase1/11 Signaling Affects Muscle Regeneration and Recovery Following Ischemia, and Can Be Modulated by Chloroquine. Mol. Med. 2020, 26, 69. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, U.; Cui, X.; Hong, G.; Namkoong, S.; Karlsson, J.M.; Baty, C.J.; Tzeng, E. High Mobility Group Box 1 Promotes Endothelial Cell Angiogenic Behavior in Vitro and Improves Muscle Perfusion in Vivo in Response to Ischemic Injury. J. Vasc. Surg. 2012, 55, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibley, R.C., 3rd; Reis, S.P.; MacFarlane, J.J.; Reddick, M.A.; Kalva, S.P.; Sutphin, P.D. Noninvasive Physiologic Vascular Studies: A Guide to Diagnosing Peripheral Arterial Disease. Radiographics 2017, 37, 346–357. [Google Scholar] [CrossRef]
- Agley, C.C.; Rowlerson, A.M.; Velloso, C.P.; Lazarus, N.R.; Harridge, S.D. Human Skeletal Muscle Fibroblasts, but Not Myogenic Cells, Readily Undergo Adipogenic Differentiation. J. Cell Sci. 2013, 126, 5610–5625. [Google Scholar] [CrossRef] [Green Version]
- Nehlin, J.O.; Just, M.; Rustan, A.C.; Gaster, M. Human Myotubes from Myoblast Cultures Undergoing Senescence Exhibit Defects in Glucose and Lipid Metabolism. Biogerontology 2011, 12, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. Aim2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih Image to Imagej: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Donato, R. Hmgb1–Rage Regulates Muscle Satellite Cell Homeostasis through P38-Mapk- and Myogenin-Dependent Repression of Pax7 Transcription. J. Cell Sci. 2012, 125, 1440–1454. [Google Scholar] [PubMed] [Green Version]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. Amphoterin Stimulates Myogenesis and Counteracts the Antimyogenic Factors Basic Fibroblast Growth Factor and S100b Via Rage Binding. Mol. Cell. Biol. 2004, 24, 4880–4894. [Google Scholar] [CrossRef] [Green Version]
- Shavlakadze, T.; Chai, J.; Maley, K.; Cozens, G.; Grounds, G.; Winn, N.; Rosenthal, N.; Grounds, M. A Growth Stimulus Is Needed for Igf-1 to Induce Skeletal Muscle Hypertrophy In Vivo. J. Cell Sci. 2010, 123, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein Hmgb1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 Initiates Apoptosis in the Absence of Gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Z.-M.; Elner, S.G.; Khanna, H.; Murga-Zamalloa, C.A.; Patil, S.; Elner, V. Expression and Functional Roles of Caspase-5 in Inflammatory Responses of Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8646–8656. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.; Bording-Jorgensen, M.; Chan, R.; Wine, E. Nigericin Promotes Nlrp3-Independent Bacterial Killing in Macrophages. Front. Immunol. 2019, 10, 2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.M.; Kang, J.H.; Yun, M.; Lee, S.B. Quercetin Inhibits the Poly(Da:Dt)-Induced Secretion of Il-18 via Down-Regulation of the Expressions of Aim2 and Pro-Caspase-1 by Inhibiting the Jak2/Stat1 Pathway in Ifn-Γ-Primed Human Keratinocytes. Biochem. Biophys. Res. Commun. 2018, 503, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Straface, G.; De Cristofaro, R.; Lancellotti, S.; Rizzo, P.; Arena, V.; Stigliano, E.; Pecorini, G.; Egashira, K.; De Angelis, G.; et al. High-Mobility Group Box 1 Protein Promotes Angiogenesis after Peripheral Ischemia in Diabetic Mice through a Vegf-Dependent Mechanism. Diabetes 2010, 59, 1496–1505. [Google Scholar] [CrossRef] [Green Version]
- Riuzzi, F.; Sorci, G.; Donato, R. The Amphoterin (Hmgb1)/Receptor for Advanced Glycation End Products (Rage) Pair Modulates Myoblast Proliferation, Apoptosis, Adhesiveness, Migration, and Invasiveness: Functional Inactivation of Rage in L6 Myoblasts Results in Tumor Formation in Vivo. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef] [Green Version]
- Chargé, S.B.P.; Rudnicki, M. Cellular and Molecular Regulation of Muscle Regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, N.; Versele, R.; de Carrizosa, M.A.D.-L.; Selvais, C.M.; Brichard, S.M.; Abou-Samra, M. Walking Down Skeletal Muscle Lane: From Inflammasome to Disease. Cells 2021, 10, 3023. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte Nlrp3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.R.; Frost, E.L.; Bellinger, C.E.; Bolte, A.; McKee, C.A.; Hurt, M.E.; Paysour, M.J.; Ennerfelt, H.E.; Lukens, J.R. Aim2 Inflammasome Surveillance of DNA Damage Shapes Neurodevelopment. Nature 2020, 580, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Loughran, P.; Shapiro, R.; Shrivastava, I.H.; Antoine, D.J.; Li, T.; Yan, Z.; Fan, J.; Billiar, T.R.; Scott, M.J. Redox-Dependent Regulation of Hepatocyte Absent in Melanoma 2 Inflammasome Activation in Sterile Liver Injury in Mice. Hepatology 2017, 65, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Matikainen, S.; Nyman, T.A.; Cypryk, W. Function and Regulation of Noncanonical Caspase-4/5/11 Inflammasome. J. Immunol. 2020, 204, 3063–3069. [Google Scholar] [CrossRef] [PubMed]
- Bitto, N.J.; Baker, P.J.; Dowling, J.; Wray-McCann, G.; De Paoli, A.; Tran, L.S.; Leung, P.L.; Stacey, K.; Mansell, A.; Masters, S.L.; et al. Membrane Vesicles from Pseudomonas Aeruginosa Activate the Noncanonical Inflammasome through Caspase-5 in Human Monocytes. Immunol. Cell Biol. 2018, 96, 1120–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolívar, B.E.; Brown-Suedel, A.N.; Rohrman, B.A.; Charendoff, C.I.; Yazdani, V.; Belcher, J.D.; Vercellotti, G.M.; Flanagan, J.M.; Bouchier-Hayes, L. Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven Il-1β Release and Cell Death. J. Immunol. 2021, 206, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal Singh, M.; Khaket, T.P.; Bajpai, V.K.; Alfarraj, S.; Kim, S.G.; Chen, L.; Huh, Y.S.; Han, Y.K.; Kang, S.C. Morin Hydrate Sensitizes Hepatoma Cells and Xenograft Tumor towards Cisplatin by Downregulating Parp-1-Hmgb1 Mediated Autophagy. Int. J. Mol. Sci. 2020, 21, 8253. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cui, X.; Li, J.; Koutakis, P.; Pipinos, I.; Tzeng, E.; Chen, A.; Sachdev, U. Chloroquine Improves the Response to Ischemic Muscle Injury and Increases Hmgb1 after Arterial Ligation. J. Vasc. Surg. 2018, 67, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Kineman, R.D.; del Rio-Moreno, M.; Sarmento-Cabral, A. 40 Years of Igf1: Understanding the Tissue-Specific Roles of Igf1/Igf1r in Regulating Metabolism Using the Cre/Loxp System. J. Mol. Endocrinol. 2018, 61, T187–T198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochat, A.; Fernandez, A.; Vandromme, M.; Molès, J.-P.; Bouschet, T.; Carnac, G.; Lamb, N.J.C. Insulin and Wnt1 Pathways Cooperate to Induce Reserve Cell Activation in Differentiation and Myotube Hypertrophy. Mol. Biol. Cell 2004, 15, 4544–4555. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, R.; Xie, B.; Assaf, E.; Morder, K.; Scott, M.; Liao, H.; Calderon, M.J.; Ross, M.; Loughran, P.; Watkins, S.C.; et al. Inflammatory Caspase Activity Mediates HMGB1 Release and Differentiation in Myoblasts Affected by Peripheral Arterial Disease. Cells 2022, 11, 1163. https://doi.org/10.3390/cells11071163
Ferrari R, Xie B, Assaf E, Morder K, Scott M, Liao H, Calderon MJ, Ross M, Loughran P, Watkins SC, et al. Inflammatory Caspase Activity Mediates HMGB1 Release and Differentiation in Myoblasts Affected by Peripheral Arterial Disease. Cells. 2022; 11(7):1163. https://doi.org/10.3390/cells11071163
Chicago/Turabian StyleFerrari, Ricardo, Bowen Xie, Edwyn Assaf, Kristin Morder, Melanie Scott, Hong Liao, Michael J. Calderon, Mark Ross, Patricia Loughran, Simon C. Watkins, and et al. 2022. "Inflammatory Caspase Activity Mediates HMGB1 Release and Differentiation in Myoblasts Affected by Peripheral Arterial Disease" Cells 11, no. 7: 1163. https://doi.org/10.3390/cells11071163
APA StyleFerrari, R., Xie, B., Assaf, E., Morder, K., Scott, M., Liao, H., Calderon, M. J., Ross, M., Loughran, P., Watkins, S. C., Pipinos, I., Casale, G., Tzeng, E., McEnaney, R., & Sachdev, U. (2022). Inflammatory Caspase Activity Mediates HMGB1 Release and Differentiation in Myoblasts Affected by Peripheral Arterial Disease. Cells, 11(7), 1163. https://doi.org/10.3390/cells11071163